Tallinna Tehnikaülikool Matemaatika-loodusteaduskond Geenitehnoloogia Instituut Molekulaar- ja rakubioloogia praktikum YTM0012 MOLEKULAARBIOLOOGIA PRAKTIKUM Pipeteerimine Automaatpipetiga pipeteerimise põhitõed Pipeteerimisel on kõige olulisem võtta õige suurusega pipett Pipeteeritav vedelik peab olema homogeenne. Seintelt ja korgi küljest tuleks tilgad ja kondensaat põhja fuugida Enne pipeteeritavasse lahusesse viimist vajuta kolb esimese astmeni alla. Pipeti
Tallinna Tehnikaülikool Matemaatika-Loodusteaduskond Geenitehnoloogia Instituut Molekulaar- ja rakubioloogia praktikum Aruanne Koostanud: Luise Tiks YASB61 112332 Tallinn 2014 Pipeteerimine Harjutus 1 1 ml pipetti kasutades pipeteerisin 15-ml falconisse 1,65 ml detergenti, 3,85 ml vett (kokku 5,5 ml 30% lahust). Detergendi pipeteerimiseks kasutasin pahupidi tehnikat.
Molekulaar- ja rakubioloogia praktikum ARUANNE Ave Tüür 155356 YAGB41 Kevadsemester 2017 Tallinna Tehnikaülikool Pipeteerimine 8. veebruar I HARJUTUS Eesmärk: Õppida õigesti pipeteerima. Materjalid: Arvutused: Pesuvahendi kontsentraat Pesuvahendit on vaja: 5,5 ×30 Vesi 100 = 1,65ml Pipetid ja otsikud 5,5−2 ×1,55=2,4 ( ml ) 15ml falkon tuub 2,4 ÷ 0,2=12 1,5 ml tuubid PCR plaadi tükk Töö käik: Teen 15-ml falkon tuubi 5,5ml 30% homogeenset pesuvahendilahust Võtan kaks 1,5ml- tuubi ning pipeteerin mõlemasse 1,55ml lah
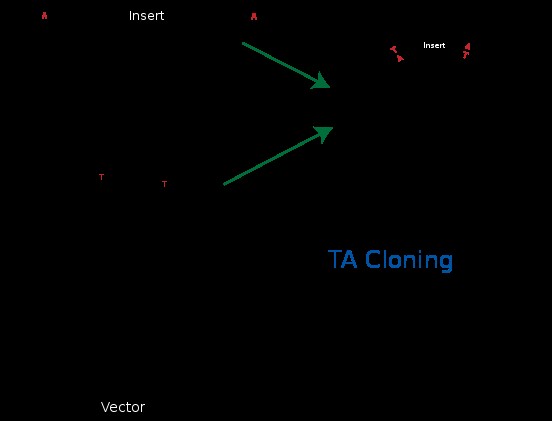
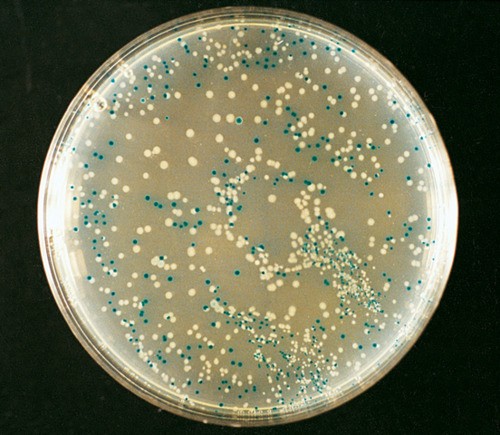
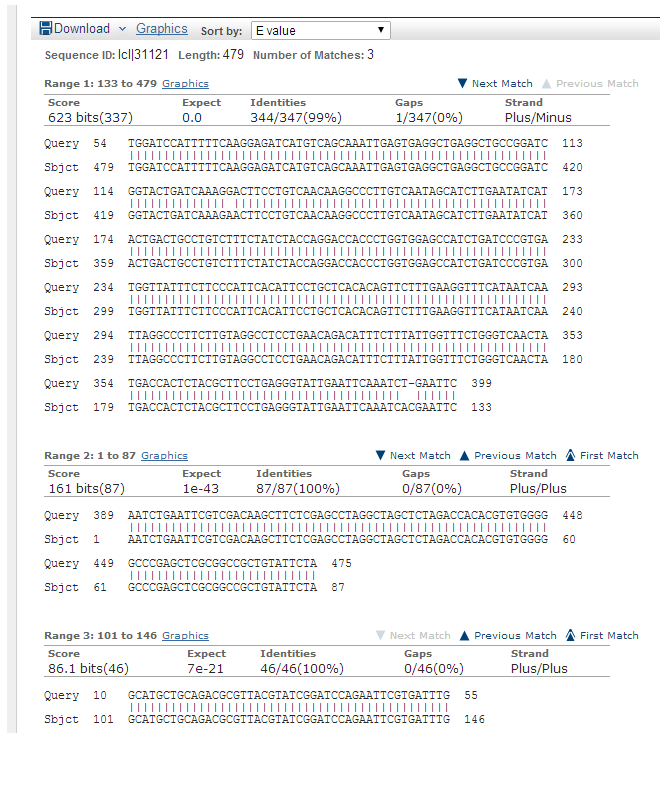
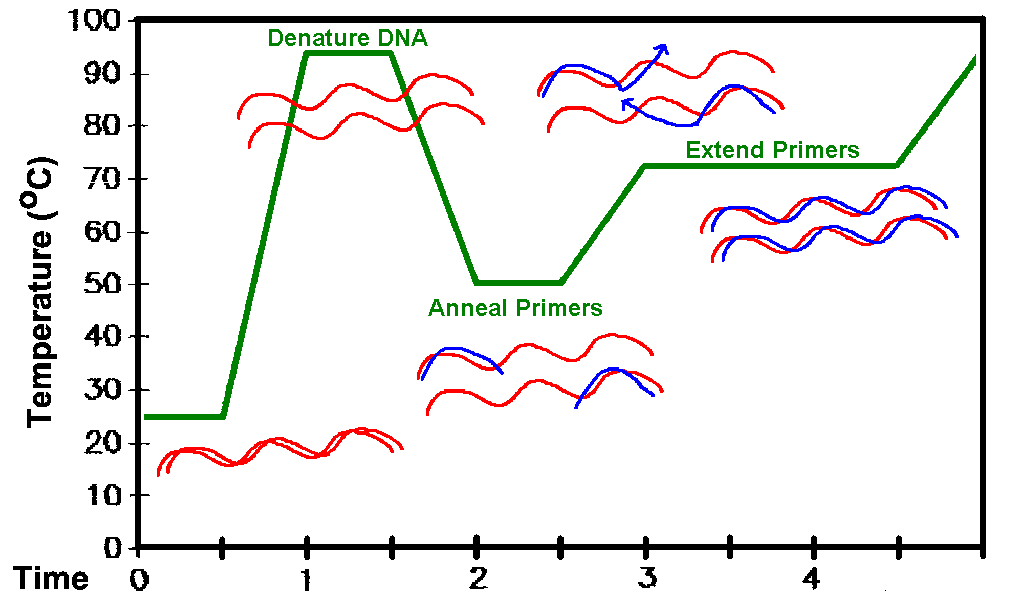
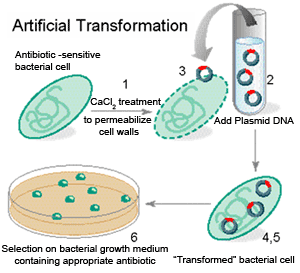
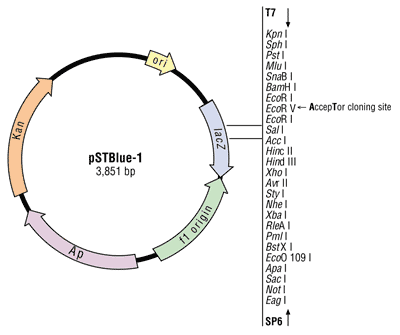
Molekulaarbioloogia I praktikumi töö (2012 a) Nimi: Inimese genoomi CpG "saarekestel" paiknevate geenide kloneerimine (genoomse DNA restriktsioon Cfr42I (SacII) abil, fragmentide kloneerimine pBS SK+ plasmiidi, E.coli transformatsioon, rekombinantse klooni eraldamine, restriktsioon-analüüs, PCR, sekveneerimine, bioinformaatiline analüüs) Imetajate genoomides on mitmed geenid koondunud nn. CpG saarekestele (CpG islands). CpG saareke on vähemalt 200 bp pikkune DNA lõik, milles on dinukleotiidi GC sisaldus vähemalt 50 %. Tavaliselt leidub CpG saarekesi (eriti koduhoidja-)geenide transkriptisooni alguspunktides või nende lähedal. Mujal genoomi piirkondades on CpG dinukleotiidi vähe, sest sellise dinukleotiidi tsütosiin metüleeritakse (replikatsioonijärgselt aitab eristada värskelt sünteesitud DNA ahelat vanast ahelast). Ekspresseeruvate geenide promootorpiirkondi ei metüleerita. Metüleeritud tsütosiin allub kergesti spontaansele de
Molekulaarbioloogia I praktikumi töö (2013 a) Nimi: YAGB41 Inimese genoomi CpG "saarekestel" paiknevate geenide kloneerimine (genoomse DNA restriktsioon Cfr42I (SacII) abil, fragmentide kloneerimine pBS SK+ plasmiidi, E.coli transformatsioon, rekombinantse klooni eraldamine, restriktsioon-analüüs, PCR, sekveneerimine, bioinformaatiline analüüs) Imetajate genoomides on mitmed geenid koondunud nn. CpG saarekestele (CpG islands). CpG saareke on vähemalt 200 bp pikkune DNA lõik, milles on dinukleotiidi GC sisaldus vähemalt 50 %. Tavaliselt leidub CpG saarekesi (eriti koduhoidja-)geenide transkriptisooni alguspunktides või nende lähedal. Mujal genoomi piirkondades on CpG dinukleotiidi vähe, sest sellise dinukleotiidi tsütosiin metüleeritakse (replikatsioonijärgselt aitab eristada värskelt sünteesitud DNA ahelat vanast ahelast).
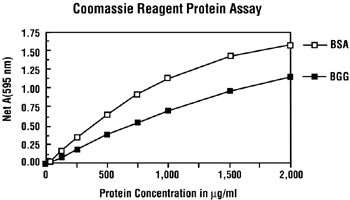
Tallinna Tehnikaülikool IMMUNOLOOGIA Praktikum Õppejõud: Lilian Järvekülg Õpilane: YAGB41 Tallinn 2012 SDS-PAGE ELEKTROFOREES · Meetod võimaldab valkude komplekside lahutamist molekulaarmassi järgi ja annab liigikaudse hinnangu valgu kontsentratsioonist. · Valgud jooksevad geelis ,,+" poole, kuna nad on kaetud SDS-ga, mis annab neile ,,- ,, laengu. Liikumine geelis toimub lahuses oleva OH- rühma tõttu. · Geelile kanname mitte ainult uuritavad valgud, vaid ka markeri, mis on kindla suurusega valkude segu (selle järgi teeme hiljem kindlaks enda uuritud valgu suuruse, kD). Töö käik: 1. Geeli valmistamine: Lahutav geel (alumine geel), Kontsentreeriv geel (ülemine 10% geel) , 4% Vesi
Tallinna Tehnikaülikool Immunoloogia Praktikum Õppejõud: Lilian Järvekülg Õpilane: Agnes Kivistik 112483YAGB42 Tallinn 2012 SDS-PAGE ELEKTROFOREES Meetod võimaldab valkude komplekside lahutamist molekulaarmassi järgi ja annab liigikaudse hinnangu valgu kontsentratsioonist. Valgud jooksevad geelis „+“ poole, kuna nad on kaetud SDS-ga, mis annab neile „-„laengu. Liikumine geelis toimub lahuses oleva OH - rühma tõttu. Geelile kanname mitte ainult uuritavad valgud, vaid ka markeri. Marker- valkude segu, mis on kindla suurusega. Töö käik: 1. Geeli valmistamine: Lahutav geel (alumine Kontsentreeriv geel geel), (Ülemine geel) , 4% 10% Vesi 4,5 ml 3 ml 4x separating(
Tallinna Tehnikaülikool Immunoloogia Praktikum Õppejõud: Lilian Järvekülg Õpilane: YAGB-42 Tallinn 2012 SDS-PAGE ELEKTROFOREES · Meetod võimaldab valkude komplekside lahutamist molekulaarmassi järgi ja annab liigikaudse hinnangu valgu kontsentratsioonist. · Valgud jooksevad geelis ,,+" poole, kuna nad on kaetud SDS-ga, mis annab neile ,,- ,,laengu. Liikumine geelis toimub lahuses oleva OH- rühma tõttu. · Geelile kanname mitte ainult uuritavad valgud, vaid ka markeri. Marker- valkude segu, mis on kindla suurusega. Töö käik: 1. Ge






































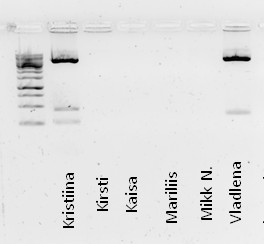
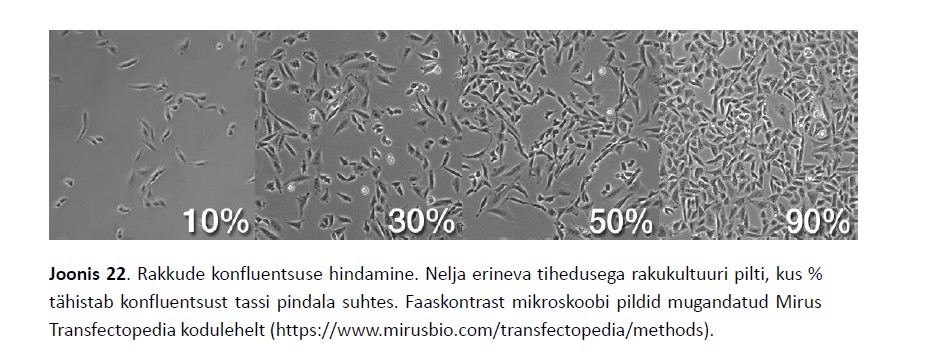
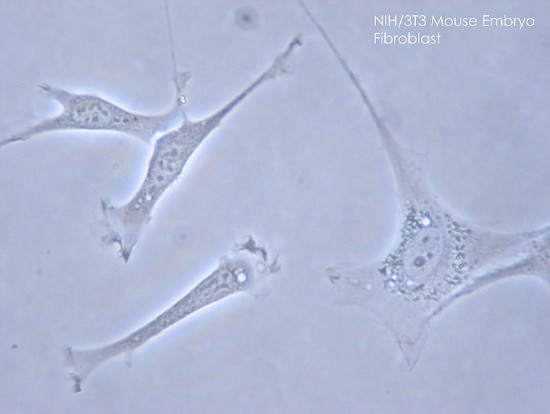
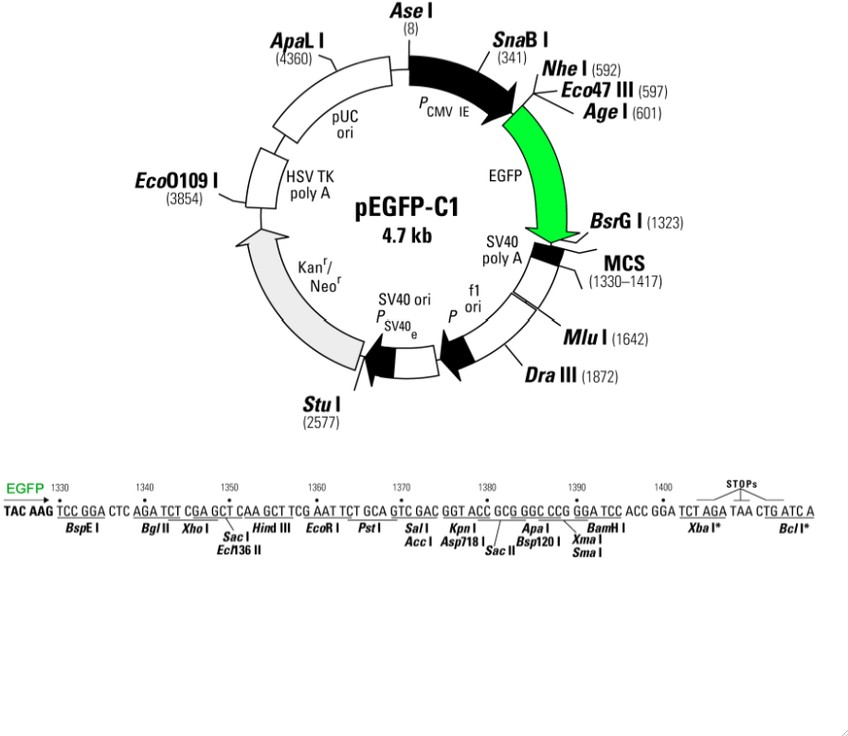

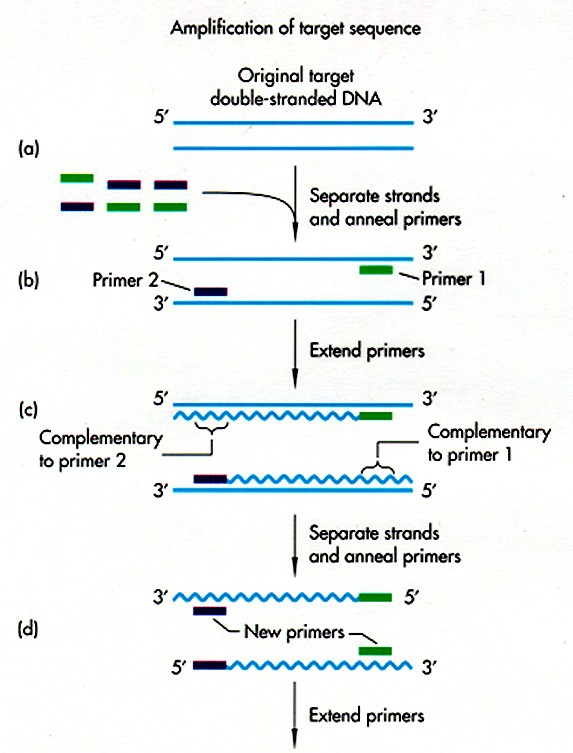


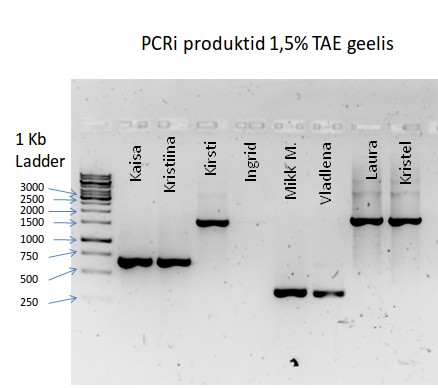
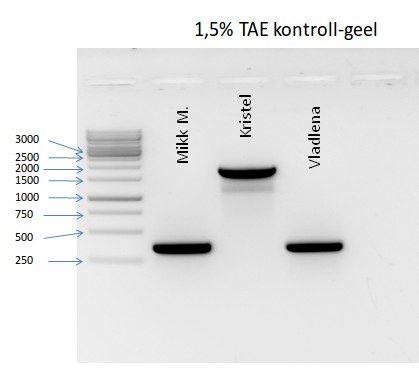




Kõik kommentaarid