Molekulaarbioloogia praktikumi aruanne YAGB41 Kevad 2014 Vlada Šiposa Molekulaarbioloogia praktikumi aruanne 1. Praktikum – pipepteerimine. Töötasime erineva suuruse pipettiga: 1000, 200, 20 ning 10 µl. Igale pipettile sobib oma suurusega otsik. Otsikud on steriilsed ja neid ei tohi neid näpuga katsuda. Pärast kasutamist tuleb otsikud ära visata spetsiaalsele konteinerisse. Otsik peab istuma tihkelt, kuna kui istub liiga lõdvelt, siis sissetõmbav kogus on tegelikult vähem, kui oodetud (tõmmatakse sisse ka õhk)
Tallinna Tehnikaülikool Matemaatika-Loodusteaduskond Geenitehnoloogia Instituut Molekulaar- ja rakubioloogia praktikum Aruanne Koostanud: Luise Tiks YASB61 112332 Tallinn 2014 Pipeteerimine Harjutus 1 1 ml pipetti kasutades pipeteerisin 15-ml falconisse 1,65 ml detergenti, 3,85 ml vett (kokku 5,5 ml 30% lahust). Detergendi pipeteerimiseks kasutasin pahupidi tehnikat.
Seega on antud töö eesmärgiks kloneerida CpG saarekestel paiknevaid geene, täpsemalt geenide regulaatoralasid ja saadud järjestuste kuuluvus teha kindlaks DNA järjestuste määramise (sekveneerimise) abil. Saadud töö tulemused võivad leida kasutust molekulaargeneetika laboratooriumi teaduslikus uurimistöös. Käesolevas töös saab üliõpilane ettekujutuse genoomse DNA restriktsioonist, kloneermisest, raamatukogu-valmistamisest, PCR reaktsioonist, DNA sekveneerimisest, jt. molekulaarbioloogia põhilistest töömeetoditest. Iga üliõpilane esitab pärast töö teostamist protokolli (kas paberil või elektroonselt), milles toob ära põhilised tulemused ja lisaülesannete vastused. Kirjelduses märgib, millised etapid läbis edukalt ja millised olid probleemsed kohad. Töö nr 1: Inimese genoomse DNA restriktsioon Cfr42I restriktaasi abil. Saadud fragmentide ligeerimine "Bluescript" pBS SK+ plasmiidi ehk vektorisse. Lähteained:
Seega on antud töö eesmärgiks kloneerida CpG saarekestel paiknevaid geene, täpsemalt geenide regulaatoralasid ja saadud järjestuste kuuluvus teha kindlaks DNA järjestuste määramise (sekveneerimise) abil. Saadud töö tulemused võivad leida kasutust molekulaargeneetika laboratooriumi teaduslikus uurimistöös. Käesolevas töös saab üliõpilane ettekujutuse genoomse DNA restriktsioonist, kloneermisest, raamatukogu-valmistamisest, PCR reaktsioonist, DNA sekveneerimisest, jt. molekulaarbioloogia põhilistest töömeetoditest. Iga üliõpilane esitab pärast töö teostamist protokolli (kas paberil või elektroonselt), milles toob ära põhilised tulemused ja lisaülesannete vastused. Kirjelduses märgib, millised etapid läbis edukalt ja millised olid probleemsed kohad. Töö nr 1: Inimese genoomse DNA restriktsioon Cfr42I restriktaasi abil. Saadud fragmentide ligeerimine "Bluescript" pBS SK+ plasmiidi ehk vektorisse. Lähteained:
Molekulaar- ja rakubioloogia praktikum ARUANNE Ave Tüür 155356 YAGB41 Kevadsemester 2017 Tallinna Tehnikaülikool Pipeteerimine 8. veebruar I HARJUTUS Eesmärk: Õppida õigesti pipeteerima. Materjalid: Arvutused: Pesuvahendi kontsentraat Pesuvahendit on vaja: 5,5 ×30
0. teema 1. Miks ei saa mikroskoobi suurendust tõsta nähtava valguse lainepikkusega sarnase suuruse objektide jälgimisel? Valguse difraktsioon piirab. Valguskiirte paindumine väikeste esemete ümber tekitab difraktsiooniringid, mis ei võimalda väikesi ja lähestikku paiknevaid objekte eraldi näha. 2. Mis on mikroskoobi lahutusvõime? Vähim punktidevaheline kaugus d, mida on võimalik mikroskoobis eristada. 3. Mis on numbriline apertuur ja millest ta sõltub? korrutis n x sinα/2. Iseloomustab objektiivi läätse võimet valgust koondada. Sõltub objektiivi ja preparaadivahelise keskkonna murdumisnäitajast (n). 4. Kuidas on võimalik mikroskoobi lahutusvõimet tõsta? Suurendada NA-d ehk keskkonna murdumisnäitajat suurendada. Optiliselt tihedama keskkonna murdumisnäitaja on vedelikul suurem kui õhul. Kasutatakse immersiooniõli preparaadi ja objektiivi läätse vahele (optiliselt tihedam keskkond). 5. Kui suur on valgusmikroskoobi lahutusvõime piirväärtus? 0,2 mikrom
Tartu Ülikool Mikrobioloogia instituut Meditsiinilise mikrobioloogia praktikum II osa Tatjana Brilene, Kai Truusalu, Tõnis Karki 2014/2015 1 Sisukord 1. Mikrobioloogilise diagnostika põhiskeem. Stafülokokknakkuste diagnostika. Streptokokknakkuste diagnostika..................................3 2. Enterobakterite nakkuste diagnostika uroinfektsioonide näitel............................................12 3. Enterobakterite nakkuste diagnostika sooleinfektsioonide näitel.........................................16 4. Bordetella ja Corynebacterium’i nakkuste diagnostika..........................................................21 5. Mycobacterium spp. infektsioonide diagnostika....................................................................26 6. Anaeroobsete infektsioonide mikrobioloogiline diagnostika.................................................32 7. Spiroheetid
B variant 1) Kirjeldage mikroorganismide külvamise tehnikaid. Millal on otstarbekas üht või teist kasutada? Joonkülvi kasutatakse põhiliselt tahketel agarsöötmetel kasvatatavate ja säilitatavate mikroorganismide kollektsioonide või töökultuuride uuendamiseks. Külvatakse enamasti katseklaasidesse valatud längagarile (kaldagarile), vahel ka Petri tassis olevale söötmele. Külvinõelaga kantakse kultuur agari pinnale (analoogiliselt joonkülviga kaldagarile) kas teatud sektorisse või üle kogu tassi pinna. Isoleeritud kolooniate saamiseks (N: puhaskultuuride eraldamisel) kasutatakse joonkülvi meetodi modifikatsiooni. Steriilse külviaasa või -nõelaga võetakse veidi külvimaterjali ja tõmmatakse sellega Petri tassi ühte serva mõned paralleeljooned (skeemil A). Külvinõel steriliseeritakse leegis ja läbi joonte A lõppude tõmmatakse ristjooned B. Külvinõel steriliseeritakse uuesti ja läbi joonte B tõmmatakse jooned C jne. Külvatava materjali k































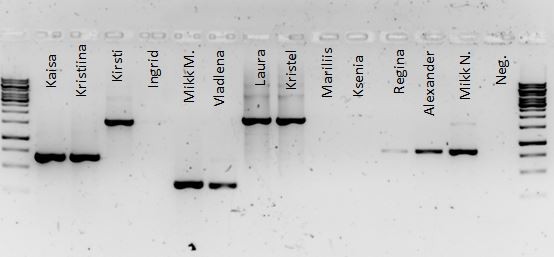
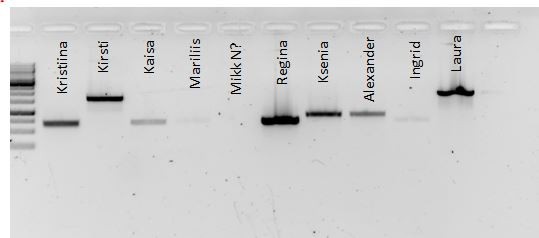
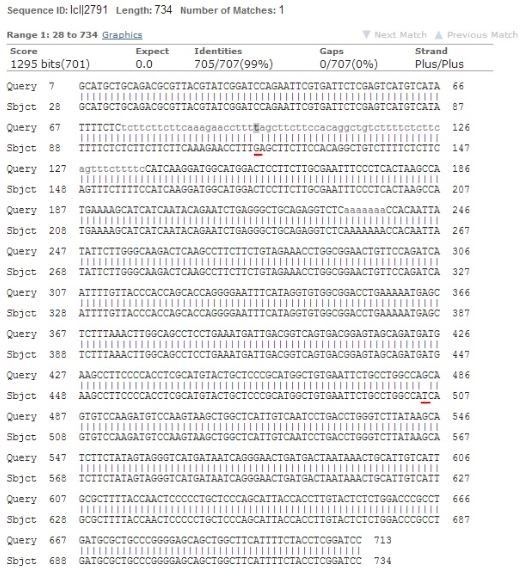




Kõik kommentaarid