Molekulaarbioloogia I praktikumi töö (2013 a) Nimi:
YAGB41Inimese
genoomi CpG “saarekestel” paiknevate geenide kloneerimine
(genoomse DNA
restriktsioon Cfr42I (SacII) abil,
fragmentide kloneerimine pBS SK+
plasmiidi,
E.coli transformatsioon ,
rekombinantse klooni
eraldamine, restriktsioon-analüüs, PCR,
sekveneerimine ,
bioinformaatiline analüüs)
Imetajate
genoomides on mitmed
geenid koondunud nn. CpG saarekestele (CpG
islands ). CpG saareke on vähemalt 200 bp
pikkune DNA lõik, milles
on dinukleotiidi GC sisaldus vähemalt 50 %. Tavaliselt leidub CpG
saarekesi (eriti koduhoidja-)geenide transkriptisooni alguspunktides
või nende lähedal. Mujal genoomi piirkondades on CpG dinukleotiidi
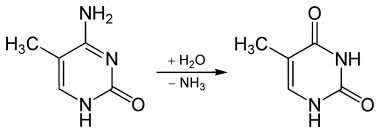
vähe, sest sellise dinukleotiidi tsütosiin metüleeritakse
(replikatsioonijärgselt aitab eristada värskelt sünteesitud DNA
ahelat vanast
ahelast ). Ekspresseeruvate geenide promootorpiirkondi
ei metüleerita. Metüleeritud tsütosiin allub kergesti spontaansele
desamineerimisele, tekib tümiin (reparatsiooniensüüm
tümiin-DNA-glükosülaas eemaldab vigasest T/G paarist T väga
väikese efektiivsusega) – seega jääb alles väga väike osa CpG
järjestustest.
Seetõttu
on CpG dinukleotiid evolutsiooniliselt säilunud ainult seal, kus
tsütosiin ei ole metüleeritud, st ekspresseruvate geenide
promootorpiirkondades.
TSÜTOSIIN 5-METÜÜLTSÜTOSIIN TÜMIIN
Selleks,
et eraldada selliseid
saari ülejäänud genoomi suuresti A-T
rikastest piirkondadest oleks sobiv kasutada restriktaasi, mis
hüdrolüüsib C-G järjestusi (näiteks Cfr42I (SacII), CCGC’GG;
NotI GC’GGCCGC;). Need restriktaasid on reeglina tundlikud C
metülatsioonile CmpG järjestuses, võimaldades
restrikteerida seega aktiivseid geene (inaktiivsed võivad olla
metüleeritud).
Seega
on antud töö eesmärgiks kloneerida CpG saarekestel paiknevaid
geene, täpsemalt geenide regulaatoralasid ja saadud järjestuste
kuuluvus teha kindlaks DNA järjestuste määramise (sekveneerimise)
abil. Saadud töö tulemused võivad leida
kasutust molekulaargeneetika laboratooriumi teaduslikus uurimistöös.
Käesolevas
töös saab üliõpilane ettekujutuse genoomse DNA restriktsioonist,
kloneermisest, raamatukogu-valmistamisest, PCR reaktsioonist, DNA
sekveneerimisest, jt. molekulaarbioloogia põhilistest
töömeetoditest.
Iga üliõpilane esitab pärast töö teostamist
protokolli (kas paberil või elektroonselt), milles toob ära
põhilised tulemused ja lisaülesannete vastused. Kirjelduses märgib,
millised etapid läbis edukalt ja millised olid probleemsed kohad.Töö
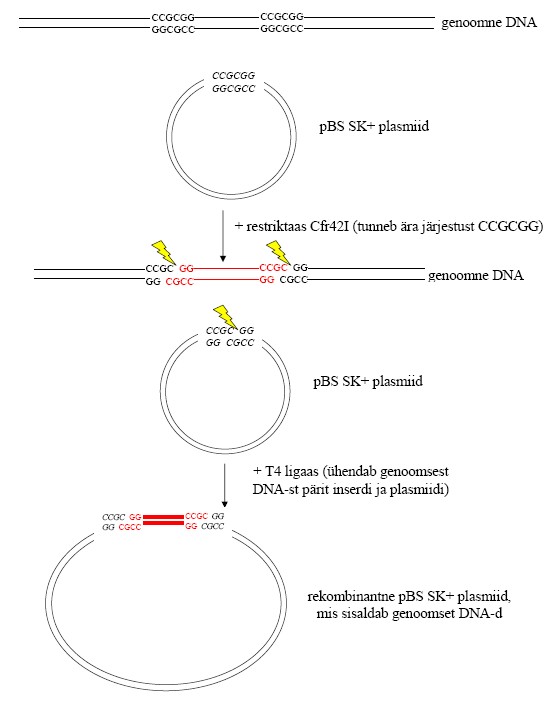
nr 1: Inimese genoomse DNA restriktsioon Cfr42I
restriktaasi abil. Saadud fragmentide ligeerimine “Bluescript”
pBS SK+ plasmiidi ehk vektorisse.Lähteained:
– Inimese
genoomne DNA ( 5 ng/μl.
(4)
Inkubeeri toatemperatuuril kuni 24 h. Reaktsiooni võib alustada ka
madalamal temperatuuril (näiteks 15 oC).
Inkubeerimisaegadel
lahendada allpool toodud lisaülesanne, vt lisamaterjale ja kuula
juhendaja näpunäiteid.
Lisaülesanded
(teoreetilised): Vaata Fermentase kataloogist, milline on Cfr42I jaoks kõige sobivam puhver (viie puhvri süsteemis). Millistes puhvrites see ensüüm veel töötab? Lisaks tutvu kataloogis olevate tingmärkidega (millisel temperatuuril ensüümid töötavad, kuidas neid inaktiveeritakse, milline on antud restriktaasiga lõigatud fragmentide ligeerimise efektiivsus jne).
Kõige
sobivamad on puhver B, G ja Tango . Ensüümid töötavad 37 kraadi
juures, inaktiveeruvad 65 kraadi juures, efektiivsus 95%, tundlik CpG
metülatsioonile.
1.2)
Kui restrikteerimiseks kasutada üheaegselt kahte erinevat
ensüümi (näiteks Cfr42I ja PstI), siis milliseid puhvreid oleks
kõige mõistlikum kasutada (vt Fermentase kataloogist restriktaaside
tabelist)?
Kas
Yello Tango puhvris oleks võimalik üheaegselt lõigata Cfr42I ja
SmaI ensüümidega?
Parim
B, sobivad ka G ja Tango. Tango puhvris on võimalik lõigata
üheaegselt nende ensüümidega, kuid kuna nad on aktiivsed
erinevatel temperatuuridel , siis on efektiivsus suhtselt madal.
1.3)
Kui palju tuleb võtta 1 M Tris-HCl, 5 M NaCl, 20% SDS ja 0,5 M EDTA -Na2 lahuseid, et valmistada 1 ml 2 x PK puhvrit (0,4
M Tris-HCl, pH 7,5; 0,44 M NaCl, 2% SDS, 25 mM EDTA-Na2).
400µl
Tris –HCl, 88µl NaCl, 100 µl SDS ja 50 µl EDTA-Na2 lahuseid
1.4)
On vaja ligeerida 100 ng 5000 bp pikkune plasmiid ja 1000 bp pikkune
insert. Mitu ng tuleb võtta inserti, et see oleks ekvivalentne plasmiidiga (arvesse tuleb võtta nende 5-kordset pikkuste
erinevust)? Mitu ng tuleks inserti võtta, kui ligeermisreaktsiooni
panna inserti 3 korda rohkem kui ekvivalentne kogus plasmiidi?
Ekvivalentne
kogus oleks 20 ng (1000bp*100ng/5000bp). Tegelikkuses peaks võtma 3x
rohkem ehk 60 ng.
Töö
nr 2: Ligeeritud produktide transformeerimine E.coli
rakkudesse.
Lähteained:
– Ligeeritud
genoomne DNA (6 μl).
– TE,
10 mM Tris-HCl, pH 7,5; 1 mM EDTA-Na2.
– E.coli
DH5 alfa kompetentsed rakud ( valmistamist vaata lisast)
– Steriilne
LB (lysogeny broth ) sööde (koostis 1 liitri lahuse kohta , 10 g
trüptooni, 5 g pärmi ekstrakti ja 5 g NaCl)
– agar
– Ampitsilliini
(Amp) vesilahus , 100 mg/ml
– IPTG (isopropüültio-beta-D-galaktosiid), 0,5 M vesilahus
– X-gal
(5- bromo -4-kloro-3-indolüül-beta-D-galaktosiid), 20 mg/ml
– 70% Etanool .
Töö
käik:
(1)
Transformatsiooniks lahjenda saadud ligeerimissegu (6 μl) TE-ga
10-30 korda (1 osa ligeerimissegu ja 9-29 osa TE-d), sega korralikult
ja pipeteeri 10 μl lahjendatud segu uude katseklaasi.
Võtsin
3 μl ligeerimissegu ja 6
μl TE-d ( tegin 3-kordse lahjenduse).
(2)
Transformatsioon:
10
μl ligeeritud DNA lahjendust 9
μl 3-kordset lahjendust
+
20 μl E.coli DH5 alfa kompetentseid rakke (hoitakse 0
oC juures)
Paiguta tuub jääle. Sega ettevaatlikult (näpuga), kompetentsed rakud on
õrnad! Inkubeeri 30 min jääl, vahete-vahel segades.
(3)
Teosta “ heat shock”, viies tuub 5 minutiks 37 oC
juurde; seejärel tagasi jääle.
(4)
Lisa 180 μl LB söödet, sega korralikult ning inkubeeri 37 oC
30 min (segamisega või ilma).
Inkubeerimise
ajal valmista LB-Ampitsillini Petri tassid (vt. p5 allpool)
(5)
Kahesajale milliliitrile LB söötmele lisa 3g Bacto-Agarit ja keeda mikrolaineahjus kuni agari täieliku sulamiseni (ettevaatust, agaroos võib kergesti üle keeda. Selleks, et seda ei juhtuks, vali sobiva
suurusega klaaskolb ja ahju reziim ning sega sagedasti). Seda tööd
teosta grupi (~10 üliõpilast) peale juhendaja juuresolekul.
Arvuta:
Kui palju on vaja võtta ampitsilliini kontsentratsiooniga 100 mg/ml
200 ml LB söötme kohta, et selle lõppkontsentratsioon oleks 100
μg/ml? 200 μl (100*200ml/100000ml)
(6) Jahuta LB-agar 50-60 oC vesivannis alla ja seejärel lisa
antibiootikum (Amp, 100 mg/ml) lõppkontsentratsioonini 100 μg/ml,
sega korralikult ning vala välja Petri tassidele
(ca
15-20 ml ühele tassile). Liiga kuumas (>60 oC) söötmes
antibiootikum laguneb! Tassid tarduvad ~15 min pärast.
Iga
üliõpilane saab ühe Petri tassi ja kirjutab oma nime plaadi äärde
(sellele poolele, kus on agar).
(7)
Transformeeritud rakkude külvamine Petri tassidele:
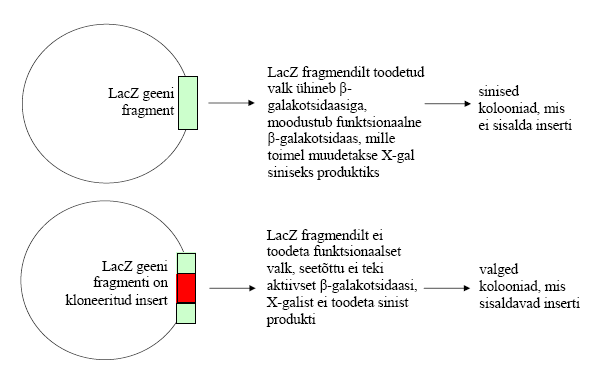
Transformeeritud
rakkude tuubi lisa 10 μl IPTG ja 10 μl X-gali lahust, sega
korralikult, kuid ära tsentrifuugi ! Nende substraatide
lisamine on vajalik beta-galaktosidaasi ekspressiooniks, mis
võimaldab identifitseerida bakteri kolooniad , mis sisaldavad
rekombinantset plasmiidi (vt lisamaterjalid).
(8)
Saadud segu (230 μl) kanna pipeti abil Petri tassile ning külva
laiali kasutades piirituslambi leegis steriliseeritud Trigalski pulka . Trikalski pulka steriliseeritakse 70% etanooli lahusega.
(9)
Paiguta saadud tassid ümberpööratult 37 oC
inkubaatorisse. Kolooniate värvusreaktsioon (sinine/valge) ilmneb
12-20 h pärast. Seejärel eemalda tassid inkubaatorist (teeb
juhendaja).
(10)
Interpreteeri tulemused (ligeerimise efektiivsus, kolooniate arv,
jne.) ja vali üks valge koloonia minipreparatsiooniks (vt. järgmine
töö). Need kolooniad tuleb 1,8 ml LB vedelsöötmesse kasvama panna
praktikumi-eelsel päeval (kontakteeru ses suhtes juhendajaga).
Lisaülesanded:
2.1)
Võrdle antud transformatsiooni protokolli (mis on kasutusel GTI
Molekulaarbioloogia laboratooriumis) DNA kokaraamatus tooduga.
Millised on erinevused?
Meie
inkubeerime 5min 37 kraadi juures, kokaraamatus 1,5min 42 kraadiga.
2.2)
Millisel juhul võib bakterikoloonia sisaldada rekombinantset
plasmiid, kuid samas koloonia värvus on ikka sinine. Kuidas määrata,
et selles koloonias on rekombinantne plasmiid?
Avatud
lugemisraam, stop- koodon puudub või kui initsiatsioon algab meie
inserdist. Määrata saab restriktsiooniga, kui
multikloneerimissaidis on restriktaas SacII.
2.3)
Mis juhtub, kui jätta Amp panemata?
Võivad
hakata kasvama muud bakterikolooniad, mitte plasmiidi sisaldavad bakterid . Meile olulised plasmiidi sisaldavad bakterid on Amp’ile
resistentsed ning suudavad selles keskkonas kasvada.
2.4)
Rakkude kompetentsus on nende võime vastu võtta
DNA-d/plasmiidi. Ligeerimissegu transformatsiooniks võiks rakkude
kompetentsus olla vähemalt 106. See tähendab, et 1
mikrogrammi plasmiidi transformatsioon annab Petri tassile 106
kolooniat. Kui rakkude kompetetnsus on 106, siis kui palju
tuleks DNA-d transformeerida, et plaadil näha 100 kolooniat? Kui 100
ng plasmiidi transformatsiooni tulemusel kasvab plaadil 1000
kolooniat, siis milline on rakkude kompetentsus?
Kui
rakkude kompetentsus on 106,
siis tuleks 0,1 ng DNA-d transformeerida, et plaadil näha 100
kolooniat. Kui 100 ng plasmiidi transformatsiooni tulemusel kasvab
plaadil 1000 kolooniat, siis rakkude kompetentsus on 104.
Töö
nr 3: Rekombinantse plasmiidse DNA minipreparatsioon.
Lähteained:
– Bakterikoloonia
(sisaldab rekombinantset plasmiidi) kasvatatud Amp-LB vedel-söötmel
12-20 h (nn. „over night ” ehk ON kultuur).
Meie
rühmas oli kolooniate arv väike, st transformatsiooni efektiivsus
oli madal. Rakud olid säilitusel kaotanud kompetentsuse.
– Lahus
I (50 mM glükoos, 25 mM Tris-HCl, pH 8,0; 10 mM EDTA- Na2)
– Lahus
II (0,1 N NaOH *, 1% SDS; * erineb sellest, mis on toodud DNA
kokaraamatus; 0,1 N lahuse kasutamine 0,2 N asemel tõstab oluliselt
DNA saagist ning võimaldab eraldada maksimaalse koguse
superspiraalset plasmiidi vormi – K. Mätlik ja M. Speek,
avaldamata andmed)
– Lahus
III (5 M KOAc), säilit 4 oC juures)
– propanool
– TE
(10 mM Tris-HCl, pH 7,5; 1 mM EDTA-Na2)
– Ribonukleaas
A TE-s (5 μg/ml,; Sigma)
– Fenool ,
tasakaalustatud TE-ga (NB! Fenooli pipeteerida ainult
kummikinnastes ja tõmbe all!).
– 70%
ja 95% Etanool.
– 1
M Na-atsetaat (NaOAc), pH 5,5 (lisatakse DNA sadestamiseks)
– Agaroos
(FMC)
– 1
x TBE foreesi puhver (TBE, 50 mM Tris-boraat, pH 8,3; EDTA-Na2)
– 1
x TBE foreesi pealekandmis-puhver (5 x TBE, 20% glütserool, 0,01%
Broomfenoolsinine)
– EtBr 5 μg/μl (pipeteerida kummikinnastega, kantserogeenne)
NB!
Enne tööle asumist lugeda läbi fenooli ja etiidiumbromiidiga
töötamise ohutuseeskiri ( http://geen.ttu.ee/?id=10705 ,
vt punkt C6. Fenooli ja etiidiumbromiidiga (EtBr) töötamine).
Töö
käik:
(1)
Vala ~1,4 ml bakteri ON kultuuri tuubi, sule tuub ja tsentrifuugi
maksimaalsel kiirusel 1 min. Tuubid asetada tsentrifuugi nii, et
tuubi ja kaant ühendav osa jääks väljapoole.
(2)
Eemalda supernatant sademe vastaspoolelt (ülemine vedeliku kiht, vt
allpool olevalt skeemilt) (maksimaalselt!)
sade
ülemine vedeliku kiht
ehk
supernatant
pipetiots
ja
suspendeeri bakterimass 200 μl Lahuses I. Sega hoolikalt (vortex),
et tekiks ühtlane suspensioon . Kasuta vajadusel pipetti.
(3)
Lisa 400 μl lahust II (rakud lüüsuvad, valgud ja nukleiinhapped denatureeritakse), ning sega ettevaatlikult “end-over-end”
stiilis (segajat mitte kasutada, see lõhub kromosomaalset DNA-d, mis
võib sel juhul hiljem kaasa sadeneda!). Aseta tuub mõneks minutiks
jääle.
(4)
Lisa 300 μl jääkülma lahust III ( valgud ja kromosomaalne DNA
sadenevad välja, plasmiid renatureerub ja jääb lahusesse). Sega
hoolikalt (nagu eelmises punktis). Hoia 2-3 min jääl.
(5)
Tsentrifuugi maksimaalsel kiirusel 5-6 min (sademeks on kromosomaalne
DNA). Samal ajal pane valmis uus tuub, millesse pipeteeri 400 μl propanooli (plasmiidse DNA & RNA sadestamiseks).
(6)
Eemalda supernatant (~900 μl) ja kanna valmispandud tuubi. Sulge tuub ja sega intensiivselt (tekib plasmiidse DNA sade, RNA jääb
osaliselt supernatanti).
(7)
Tsentrifuugi maksimaalsel kiirusel 8-10 min. Eemalda supernatant
täielikult ja lahusta sade 100 μl TE + Ribonukleaasi A (5 μg/ml)
lahuses (RNA lagundamiseks). Inkubeeri saadud lahust 37 oC
juures ~30 min. Vt lisaülesanne 3.2.
1)
suurem osa 1000 μl
pipetigakiirtsentrifuug
2)
väiksema pipetiga eemaldasin ülejäänu supernatandi
(8)
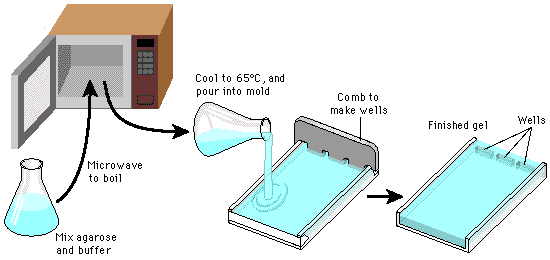
Samal ajal valmista 1 x TBE 0,8% agaroosgeel DNA analüüsiks (grupi
peale).
Selleks
on vaja 80 ml TBE puhvrit ja ... g agaroosi. Keeta mikrolaineahjus kuni agaroos on lahustunud. Jahutada vesivannis ja
lisada DNA nähtavaks tegemiseks 1 μl EtBr (5 μg/μl). Valada geel vormi, kuhu on varem valmis pandud nn „ kamm “ või „hambad“.
~30 min pärast on geel tardunud .
(9)
Peale 30 min inkubatsiooni teosta DNA-le fenooltöötlus. See on
vajalik RNaasi eemaldamiseks. Selleks:
-
lisa RNaasA-ga töödeldud DNA-le võrdses koguses fenooli (100 μl)
(tõmbe all!!!)
-
tsentrifuugi 1-2 min
-
kanna vesikiht uude tuubi (~100 μl). Vesikihis on DNA ning valgud
(RNaas) jäävad fenoolkihti ja vahekihti
-
DNA sadestamiseks lisa vesikihile 1/20 osa (5 μl) NaOAc ja 3 mahtu
(300 μl) 95% etanooli
-
sega korralikult läbi
-
tsentrifuugi maksimaalsel kiirusel 8-10 min
-
eemalda supernatant
-
pesemiseks lisa sademele ~200 μl 70% etanooliga ja tsentrifuugi
uuesti 2-3 min
DNA
Eemalda
pesulahus ja lahusta saadud DNA (peab olema silmaga nähtav kogus!)
50 μl TE-s. Selliselt eraldatud/ puhastatud DNA on valmis
restriktsioon-analüüsiks, PCR reaktsiooniks ja sekveneerimiseks
(järgmises praktikumis ).
Ei
teinud, sest lahus oli piisavalt puhas.
(10)
Kanna geelile 1/30-1/50 oma DNA lahust:
2-3
μl DNA-d 2,5 μl
DNA-d
+
10 μl 1 x TBE foreesi pealekandmis-puhver (lilla)
Pipeteeri
kogu lahus geelil olevasse „hambasse” ja elektroforeesi ~1 tund
100V juures.
Plasmiidi
eraldamine õnnestus, alumine pilv on RNA jääk, mida ei tohiks olla puhtuse mõttes.
Lisaülesanded:
3.1)
Võrdle antud protokolli DNA kokaraamatus tooduga (vt. lisa).
Millised etapid on samad, millised erinevad? Sinu arvamused,
ettepanekud, küsimused?
Punktis
7 pestakse lahust 70% etanooliga.
3.2)
Kui palju tuleb võtta RNaasA-d, mille algkontsentratsioon on 10
mg/ml, et selle lõppkontsentratsioon oleks 5 ng/μl 1 ml-s TE-s?
Mitu korda tuleb RNaasi lahjendada?
On
vaja võtta 1ml RnaasA-d, 2000x.
3.3)
Millise laenguga on DNA? Kas geeli „hammastega“ ots (millesse on kantud DNA) tuleb asetada foreesivanni + või – elektroodi poole.
DNA
on negatiivse laenguga, tuleb asetada – eletroodi poole.
3.4)
Milleks kasutatakse geelelektroforeesil etiidiumbromiidi (EtBr)?
Kuidas EtBr toimib?
DNA
nähtavaks tegemiseks (EtBr interakteerub lämmastikaluste vahele
ning hiljem UV-valguses fluorestseerub), vähendab DNA laengut (DNA
muutub pikemaks ja jäigemaks).
3.5)
Mitu grammi agaroosi on vaja 40 ml 1,5 % geeli tegemiseks? 80 ml 0,8
% geeli tegemiseks?
Et
valmistada 40 ml 1.5% geeli on vaja 0,6 g agaroosi ja , et valmistada
80 ml 0,8% geeli on vaja 0,64 g agaroosi.
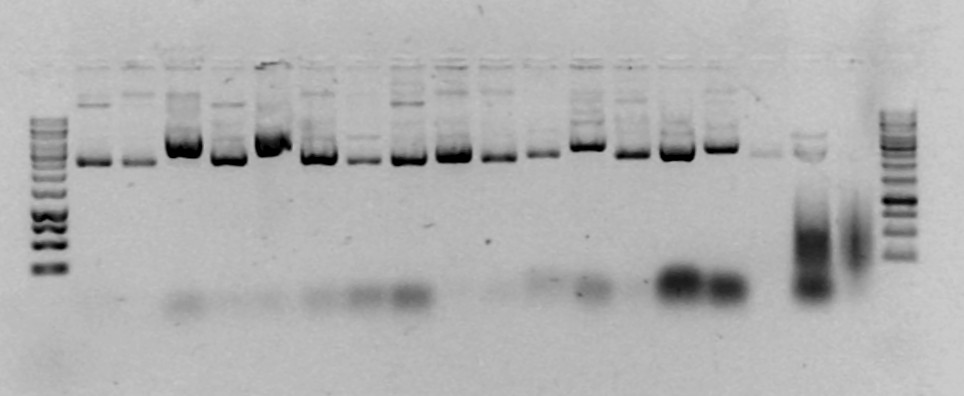
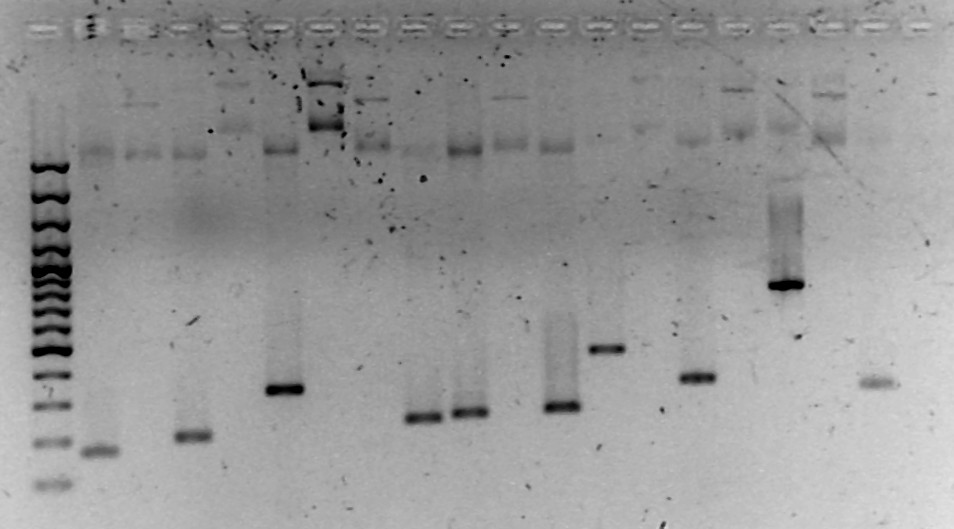
Töö
nr 4: Rekombinantse plasmiidi inserdi suuruse määramine
restriktsiooni ja/või PCR abil
(kumba
meetodit kasutada, otsusta eelmise töö põhjal, hinnates inserdi
ligikaudset suurust)
Kasutame
PCR-i. Restriktsiooni ei saa kasutada, sest meie inserdid on































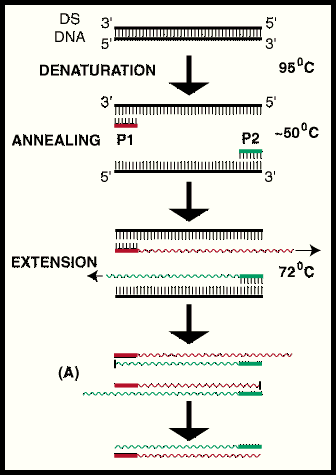




Kõik kommentaarid