Tallinna Tehnikaülikool
Matemaatika-Loodusteaduskond
Geenitehnoloogia Instituut
Molekulaar- ja
rakubioloogia praktikum
Aruanne
Koostanud :
Luise Tiks
YASB61
112332
Tallinn 2014
Pipeteerimine
Harjutus 1
1 ml pipetti kasutades pipeteerisin 15-ml falconisse 1,65 ml
detergenti, 3,85 ml vett (kokku 5,5 ml 30% lahust).
Detergendi pipeteerimiseks
kasutasin pahupidi
tehnikat . Pipeteerisin kahte
eppendorfi 1,55 ml lahust, allesjäänud 2,4 ml pipeteerisin 200 μl
kaupa eppendorfidesse – sain 11 ja pool eppendorfi.
Harjutus 2
Hindasin silmaga vee koguseid:
1. 200 μl
2. 60 μl
3. 100 μl
4. 20 μl
5. 7 μl
6. 90 μl
7. 40 μl
8. 150 μl
Hinnangud polnud küll päris täpsed, aga siiski küllaltki
lähedased.
Harjutus 3
Pipeteerisin laual oleva plaadi
viide auku 100 μl broomfenoolsinist,
lisasin vastavalt 50 μl, 5 μl, 1 μl ja 0,5 μl. Lisaks lisasin
juhendist erinevalt ühte auku 20 μl, ning pipeteerisin uue puhta
broomfenoolsinise kuuendasse auku. Tulemuseks oli küllaltki sujuv
värvirida.
Blotipaberile oma nime ja mustrit pipeteerides jälgisin, et
pipett ei läheks liiga sügavale lahusesse, et pipett oleks otse ning
pipeteerimine
toimuks sujuvalt, et
kogused oleks võimalikult täpsed
ning täpid tuleksid ühtlased.
1. Polümeraasi ahelreaktsiooni
teostamine a) Paljundamiseks sain plasmiidi pEGFP-FoxO3a-wt, praimeritest
kasutasin:
- EGFP-3-s-170-F (5’-CATGGTCCTGCTGGAGTTCGTG-3’)
- SV40p -R (5’-GAAATTTGTGATGCTATTGC-3’)
Praimerid on valitud
selliselt , et nad ei moodustaks omavahel dimeere
(ehk ei oleks omavahel komplementaarsed) ning oleksid
komplementaarsed lõiguga paljundataval DNA-l, millega praimerid
seonduvad ning kust algab
replikatsioon .
b) Kasutatud PCR-i programm:
1. 95 o C 15 minutit – ensüümi (polümeraasi)
aktiveerimiseks
2. 95 o C 10 sekundit –
denatureerimine ehk
ahelate lahku
kuumutamine 3. 56 o C 20 sekundit – praimerite hübridiseerumine
DNAle (annealing)
4. 72 o C 3 minutit – komplementaarse DNA ahela süntees
(primeri ekstensioon)
5. Korda alates 2. punktist veel 21 korda
Esimese etapi pikkus sõltub valitud ensüümist, me kasutasime Hot
FIREPol DNA polümeraasi, mis
aktiveerub pärast 15 minutit
inkubeerimist 95 o C juures. Teine etapp on vajalik
selleks, et kas lisatud template DNA või eelmises tsüklis
sünteesitud DNA
ahelad denatureeruks, see peab olema võimalikult
lühike. Kolmanda etapi (annealingu) pikkus sõltub kasutatud
praimerite järjestusest – mida suurem on G-de ja C-de arv, seda
rohkem on vaja aega annealinguks. Kuna me kasutasime erinevaid
praimereid, kuid sama programmi, ei pruugi annealingu aeg olla
täiesti optimaalne, kuid siiski piisavalt täpne. Sünteesi pikkus
sõltub nii kasutatavast ensüümist kui ka paljundatava järjestuse
pikkusest, kuna me kasutasime kõik sama programmi, siis on valitud
pigem pikem aeg (valitud on 3 minutit, lühemate järjestuste puhul
piisaks ka 1 minutist). Korduste arv sõltub sellest, kui palju on
vaja DNA-d saada, samas liiga palju tsükleid suurendab
vigade tõenäosust.
c)
Geelipildilt on näha, et PCR on tõenäoliselt ebaõnnestunud, kuna bändi pole
näha (peaks olema noolega märgitud kohas). Põhjuseks võib olla
ebatäpsus. Edasiseks tööks sain L-Envo pEGFP-C2.
PCR-i produkti puhastamine
a) Kaalusime 1,5g agaroosi, viisime mahu TAE puhvriga 100ml-ni ja
kuumutasime segu mikrolaineahjus , kuni agaroos täielikult lahustus.
Seejärel jahutasime lahust ringjate liigutustega loksutades. Jahtunud segule lisasime 0,5μl EtBr , loksutasime ning valasime
lahuse geelialusele. Lasime geelil tarduda.
b) Kuna produktide pikkus varieerus 300-2000 bp vahel, siis
valmistasime 1,5% agaroos geeli. Mida pikemad produktid , seda lahjem
peab olema geel, ning vastupidi.
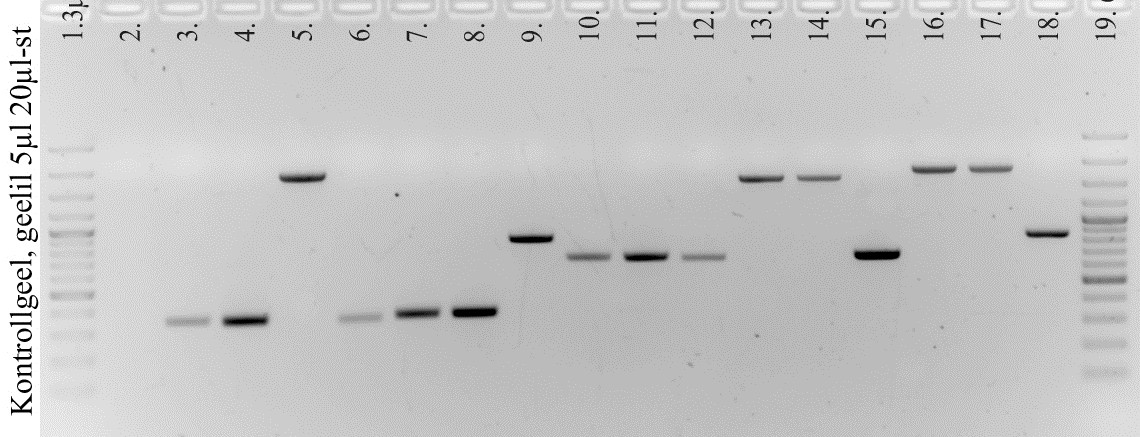
c) Kontrollgeeli
valasime selleks, et hinnata, kas puhastamine on olnud efektiivne.
Kui segusse on jäänud peale soovitava produkti muid jääke, on
kontrollgeeli pildil näha peale õige pikkusega lõigu ka teisi
bände. Kui aga geelil õige pikkusega bänd puudub, võib arvata, et
puhastamise käigus on PCR-i produkt kadunud. Minu proov asub
kolmandas positsioonis. Kontrollgeelilt on näha üks õige pikkusega
(um 300 bp) lõik, mis näitab, et puhastamine on õnnestunud.
Geeli analüüsil selgus, et inserdi kontsentratsioon oli umbes 13,7
ng/μl. Kuna vektori ja inserdi pikkuste suhe 3851/330=11,7. Kuna vektorit oli 9 ng, saame leida vektori ja inserdi koguste suhte:
(13,7*11,7)/9=17,70. See suhe on küllaltki sobiv.
Rekombinantse plasmiidi ligeerimine
Kasutasime pSTBlue-1 vektorit, mis sisaldab kahte replikatsiooniks vajalikku origini (üks kummaski suunas), ampitsilliini ja kanamütsiini resistentsusgeeni ja lacZ operoni, mille lugemisraami sisestame oma inserdi. See võimaldab sini-valge meetodil välja selgitada, millistes kolooniates on tühi, millistes meie inserti sisaldav plasmiid . Vektoril on T-overhangidega otsad , mis võimaldab A-overhangidega inserti sisestada.
PCR-i produkti on võimalik vektorisse ligeerida, kuna vektoril on T-overhang otsad ning PCR-i produktil on A-overhang otsad. T ja A on omavahel komplementaarsed, seega vektori ja inserdi „kleepuvad otsad” ühendatakse komplementaarsusprintsiibi alusel kõigepealt vesiniksidemetega, seejärel saab ligaas sünteesida fosfodiestersidemed. Polümeraasi valikul tuleb silmas pidada, et tegemist oleks Taq Polümeraasiga, mis lisab suure tõenäosusega 3’ adeniini aluse, mis on vajalik T-A kloneerimiseks.
Vektoril on kaks antibiootikumiresistentsust, seega rakke, kuhu plasmiid on transformeeritud, saab kasvatada nii ampitsilliini, kanamütsiini kui ka mõlemat sisaldavad keskkonnas – seeläbi saab vähendada ohtu, et plasmiidi mitte sisaldavad rakud tekitavad saaste.
Vektor sisaldab lacZ operoni, mis võimaldab kasutada inserti
sisaldavate rakukolooniate tuvastamiseks sini-valge meetodit.
Lisaks sellele on vektoris palju erinevaid restriktsioonisaite, see
võimaldab vektorisse sisestada erinevate restriktaasidega töödeldud
otstega inserte.
Plasmiid sisaldab lacZ operoni, mis kodeerib β-galaktosidaasi. Kui kasvusöötmele lisada X-gali (laktoosi analoog , kus galaktoosiga on glükoosi asemel seotud inool), siis rakkudes, kus lacZ lugemisraam on terve, hüdrolüüsib β-galaktosidaas X-galis glükosiidsideme, vabanevad galaktoos ja 5-bromo-4-kloro-3-hüdroksüindool, millest viimane dimeriseerub ja oksüdeerub – tekib indigosinist värvi dikloro-dibromo- indigo , mis värvib indigosiniseks terve koloonia.
Kui lacZ operoni sisestada insert , siis see rikub (suure
tõenäosusega) lacZ operoni lugemisraami, β-galaktosidaasi ei
ekspresseerita, eelkirjeldatud protsessi ei toimu ning kolooniad jäävad valged.
See võimaldab vaatluse teel eristada kolooniaid , kus insert on
plasmiidi sisenenud, ja kolooniaid, kus on vaid tühi plasmiid.
Ligeerimisprotokoll:
Pipeteerida ligeerimissegu (kokku 5 μl):
1 μl puhastatud PCR produkti
1 μl plasmiidi (9ng/ μl)
0,5 μl 10x ligeerimispuhvrit
0,5 μl e. 5 ühikut T4 DNA ligaasi (5 U/μl)(hoida jääl või
külmakehas)
2 μl Milli-Q vett
2. Homogeniseerida lahus pipeteerimise või vortexi teel (kasutasin
pipeteerimist).
3. Vajadusel tsentriguugida segu põhja (kuna kogu ligeerimissegu
pipeteerisin põhja ühte tilka kokku ning vortexit ei kasutanud,
polnud vajadust tsentrifuugimiseks).
4. Ligeerida +4oC juures üleöö.
Transformeerimine
Kompetentseteks nimetatakse rakke, mis on võimelised sisse võtma DNA-d. Kompetentsus võib olla kas loomulik või kunstlik, saavutatud laboritingimustes. Kompetentsus on vajalik selleks, et transformeerimisel soovitav plasmiid rakkudesse viia. Kompetentsete rakkude saamiseks laboritingimustes töödeldakse rakke kas KCl, CaCl või RbCl-ga, mis muudab rakumembraani DNAd läbilaskvaks.
Rakkude kompetentsuse hindamiseks tranformeeritakse 100 μl rakususpensiooni 10 pg, 100 pg ja 1 ng plasmiidiga (sobib lihtsalt tühi vektor, peaasi et kontsentratsioon on täpne). Kolooniate arvu järgi saav välja arvutada, mitu kolooniat tekib 1µg plasmiidi kohta. Kui 1 ng-ga transformeeritud tass annab 100 kolooniat, siis 1µg annab 100 000 (mis on suhteliselt viletsad). Head kompetendid on näiteks 108, see tähendab, et 1ng-ga transformeeritud tass annab 100 000 kolooniat, mida on ilmselgelt ülelugemiseks liiga palju, siis kasutatakse transformeerimisel veel väiksemaid koguseid (juba 10pg plasmiidiga transformeerimisel tekib 1000 kolooniat).
Transformeerimise protokoll :
1. Inaktiveeri ligaas inkubeerides eppendorfi 10 min 65 °C –
vajalik, et lõpetada vektori ja inserdi ligeerimine ja hoida ära
soovimatud ligeerimisprotsessid segus.
2. Transformeerimiseks võta -80°C juurest 10 minutiks jääle
sulama kompetentsed rakud. – jääs hoidmine on vajalik, kuna
külmutatud rakud on väga tundlikud kõrge temperatuuri suhtes.
3. Transformeermiseks võta 20 μl kompetentseid rakke ning lisa
ligatsioonisegule.
4. Inkubeeri 30 min jääl. – vajalik efektiivsuse suurendamiseks keemiliselt kompetentseks muudetud rakkude puhul, kuna
külmades tingimustes kinnitub DNA rakuseinale. Etapi võib jätta
vahele, või lühendada, kuid see vähendab transformatsiooni efektiivsust .
5. Selle aja jooksul vala LB+Amp+X-gal+ IPTG tassid
6. Heat shock: inkubeeri transfornatsioonisegu 90 sekundit 42oC
juures. – järsk temperatuuritõus tekitab plasmamembraani
poorid ning võimaldab DNA-l rakkudesse siseneda.
7. Tõsta tuub vähemalt 2 minutiks jääle – võimaldab
rakkudel pärast Heat-shocki rakumembraani taastada.
8. Lisa 500 μl LB vedelsöödet (sega korralikult)
9. Inkubeeri 15 min 37oC termostaadis – vajalik
selleks, et rakkudel oleks aega pärast transformatsiooni kosuda ja
hakata kiiremini paljunema. Etapid 8-11 võib ka vahele jätta, ning
külvata rakud vedelsöötme asemel otse plaadile .
10. Tsentrifuugi bakterid põhja 3000 rpm 2 minutit
11. Nüüd vala sööde (hoogsalt) rakudest moodustunud sademelt ära
kraanikaussi (tuubi jääb alles umbes 100 μl)
12. Suspendeeri pipetiga bakterid söötmesse ja kanna pipeti abil
tardunud söötmega Petri tassile
13. Kalla kuulikesed tassile (ca. 10), pane kaas peale ning loksuta
kuulikeste abil bakterid mööda tassi ühtlaselt laiali kuni vedelik
on imendunud. Vala kasutatud kuulikesed kogumisnõusse
(korduvkasutatavad).
– vajalik, et rakud plaadil ühtlaselt jaotuksid ning tekiksid
eraldatud kolooniad, mitte kõik rakud ei kasvaks plaadil üheks
suureks kolooniaks.
14. Aseta tassid inkubaatorisse üleöö kasvama. Juhendaja võtab
tassid välja 18h möödudes ning säilitab neid +4°C juures
järgmise nädalani (bakterid tassidel püsivad külmas
kasutamiskõlblikud kaks nädalat). – vajalik, et rakkudel oleks
piisavalt aega paljuneda ja kolooniad moodustada. Külma panek on
vajalik, et kogu plaat ei kasvaks ühtlaselt rakke täis ning oleks
võimalik kolooniaid eristada.
15. Päev enne järgmist praktikumi tuleb panna saadud valged
kolooniad 1 ml Amp-i
sisaldavasse vedelsöötmesse kasvama. Selleks kasutada 1,5 ml
eppendorfi, mille kordi sisse torgata nõelaga õhuauk. Tuubid tõsta
loksutavasse 37oC inkubaatorisse otimaalselt 16 tunniks
kasvama. – Vajalik selleks, et välja valida valge koloonia
rakud (rakud, millesse on sisenenud inserti sisaldav plasmiid) ning
neid paljundada.
Sulatasime mikrolaineahjus 200ml LB+ Agar söödet kuni agari täieliku sulamiseni. Täielikult üles sulatatud söötme jahutasime ~50°C juurde (kõrgemal temperatuuril võib antibiootikum hävida) ning lisasime 200 μl X-gal (lõppkonts. 20μg/ml) – vajalik sööde sini-valge selektsiooni läbiviimiseks, 25 μl IPTG-d (lõppkonts. 0,1mM) – vajalik lacZ promootori aktivatsiooniks, 200 μl ampitsilliini (lõppkonts. 100 μg/ml) – vajalik selleks, et plasmiidi mittesisaldavad rakud ei saaks plaadil kasvama hakata, ja loksutasime segamini . Saadud sooja homogeense söötme valasime õhukese kihina Petri tassidele ning jätsime tarduma.
Antibiootikumina kasutasime ampitsilliini, kuna transformeeritavas
plasmiidis on ampitsilliini resistentsusgeen, mis võimaldab
rakkudel, milles transformeerimine on õnnestunud, kasvama ja
paljunema hakata. Oleks saanud kasutada ka kanamütsiini, kuna
plasmiidis on ka kanamütsiini resistentsusgeen, kuid ampitsilliini
kasutamine on vähem komplitseeritud .
DNA minipreparatsioon:
Bakterikoloonia on bakterite kogum, mis tekib ühe bakteriraku paljunemisel.
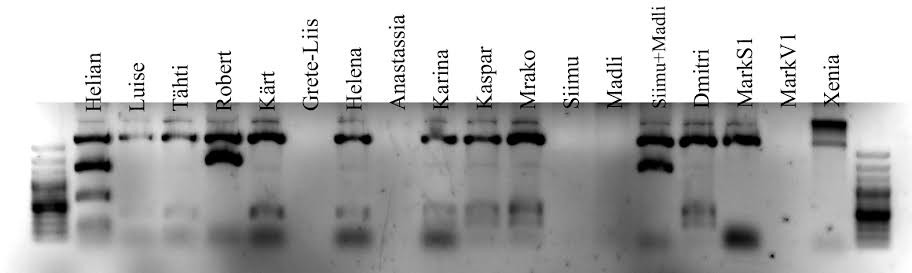
Petri tassil oli 25 kolooniat, neist 4 valged. Vedelsöötmesse panin kasvama ühe valge koloonia, kuna koloonia valge värv näitab, et nendes rakkudes on plasmiid, milles lacZ operoni lugemisraam on inserdi poolt rikutud – vastasel korral sünteesitaks rakkudes dikloro-dibromo-indigod ning kolooniad oleks sinised.
Minipreparatsiooni protokoll:
1. Võtta 1,5 ml epsis üleöö kasvanud bakterikultuur
2. Tsentrifuugi bakterid põhja 2 min maksimumpööretel
toatemperatuuril
3. Eemalda ettevaatlikult pipetiga sööde ( supernatant ehk selge
lahus sademe kohal) – eppendorfi põhja on kogunenud rakusade.
4. Re-suspendeeri pellet (väike hunnik rakke epsi põhjas) 0,2 ml-s
E1 lahuses Ole hoolas: lahusesse jäävad klämbud ei lüüsu
järgmises etapis täielikult ja seega saad vähem plasmiidi. Glükoos
muudab E1 lahuse isotooniliseks, EDTA seob kahevalentsed katioone
(seega inhibeerib paljusid ensüüme, kes töötamiseks neid enda
koostisse vajavad) ja Tris on puhverdav reagent. RNAas A eemaldab
bakteraalse RNA prepsist(ehk prepareeritavast plasmiidsest DNAst )
5. Lisa 0,2ml E2 lahust ning pipeteeri mitu korda edasi-tagasi.
E2-s sisalduv SDS solubiliseerib ( lahustab pisikesteks puhvris
ujuvateks mullikesteks e. mitsellideks ) bakteriraku membraani
fosfolipiidid ning denatureerib valgud . Naatriumhüdroksiid aga
denatureerib DNA struktuurid .
Lahus muutub“tatiseks“
6. Inkubeeri laua peal 3 minutit. Selle aja jooksul muutub lahus
selgemaks. Juhul, kui inkubeerida üle 5 minuti, saad saagise hulka
bakteri genoomseid järjestusi ning vähenab plasmiidse DNA hulk, mis
korrektselt järgmises etapis renatureerub (puhastamise saagis
väheneb!).
7. Pipeteeri peale 0,3 ml E3 lahust ja raputa segu kuni see on täiest
ühtlane.
Peaksid nägema helbeid , mitte valkjaid triipe lahuses.
8. Tsentrifuugi 10 min maksimumpööretel toatemperatuuril (RT) –
eppendorfi
põhja tekkis purune sade, vähesel määral oli valgeid helbeid
ka lahuse pinnal.
9. Tõsta supernatant uude epsi ning lisa sinna 0,5 ml (0,7 mahtu)
isopropanooli ja sega hoolikalt.
10. Tsentrifuugi plasmiidne DNA põhja 10 minutit maksimumpööretel
(RT) Pane epsi „sabad“ tsentrifuugis suunaga väljapoole, see on
oluline selleks, et teaksid kuhu pisike DNA sade tekib. – DNA
sade oli mööda eppendorfi seina umbes 0,75 ml kriipsuni
11. Eemalda ettevaatlikult supernatant.
12. Pese sadet sooladest vabanemiseks 70% etanooliga. See tähendab:
pipeteeri sademele 200μl 70% EtOH, tsentrifuugi 5 minutit
maksimumpööretel RT ning eemalda supernatant. Etanool lahustab
sademest üles DNAga kaasa tulnud mitmesuguseid soolasid.
Ettevaatust! Jälgi et DNA sade etanooli äravõtmisel kaasa ei
tuleks!
13. Kuivata DNA sade läbipaistvaks, et vabaneda ka etanooli
jääkidest.
– kuivatamise kiirendamiseks panime eppendorfis +37 kraadi
inkubaatorisse
14. Suspendeeri 30-50 μl MQ-s – DNA sade lahustus kiiresti
Edukaks DNA eralduseks peab olema transformatsioon edukas ning kasutatavate lahuste kogused ning tsentrifuugimise ja inkubeerimise ajad olema optimaalsed, igasugune kõrvalekalle vähendab saagist või eraldatud DNA puhtusastet. Saagise kogust mõjutavad tegurid:
rakkude vedelsöötmes kasvatamise aeg (optimaalne on 16h, kui kasvatada liiga vähe, ei jõua rakud paljuneda, kui liiga kaua, võib tekkida saaste või võivad rakud DNA rakkudest väljutada)
rakusademe E1 lahuses suspendeerimise hoolikus (kui sade pole täielikult lahustunud, ei lüüsu segu täielikult
E2 lahusega inkubeerimise aeg (kui inkubeerida liiga vähe, ei jõua rakud lüüsuda ning saagis jääb väike, kui aga liiga kaua, siis lisandub saagisele bakteri genoomseid järjestusi ning väheneb renatureeruva plasmiidse DNA kogus
hoolikus supernatandi eemaldamisel (tuleb eemaldada võimalikult suur kogus supernatanti, ilma, et sade kaasa tuleks, vastasel juhul kas kaotatakse osa saagisest, või võetakse kaasa jääke)
Kontroll- restriktsioon
Kontroll-restriktsiooni on vaja, et kontrollida, kas saadud plasmiid on õige, see võimaldab ka näha, kas puhastamine oli efektiivne – foreesipildilt on näha, kui proovis on lisaks saadud plasmiidile ka näiteks bakteriaalset DNA-d. Valides sobivad restriktaasid, on võimalik kontrollida, kas insert on plasmiidi sisenenud õiget pidi.
Kasutasime restriktaasi EcoRI , mis lõikab kahelt poolt EcoRV lõikamissaiti, kuhu sisestasime oma inserdi. See võimaldab tükkide pikkuste järgi määrata, kas insert on vektorisse sisenenud või mitte.
Protokoll:
Pipeteerida:
5,9 μl vett
3 μl just puhastatud plasmiidset DNA-d
1 μl restriktaasi 10x puhvrit
0,1 μl EcoRI restriktaasi (hoida külmas!)
Segada, fuugida ja inkubeerida 37oC 30 min
Valada 100 ml TAE geeli – kontsentratsioon valida oodatavate produktide suuruse järgi
Kui 30 minutit on täis, tsentrifuugi uuesti oma kontrollrestriktsiooni, et kokku koguda kaanele kogunenud veetilgad
Lisa 2 μl 6x DNA värvi ning pipeteeri segu agaroosgeeli hambasse
10-15 minuti järel tee geelist pilt ning analüüsi
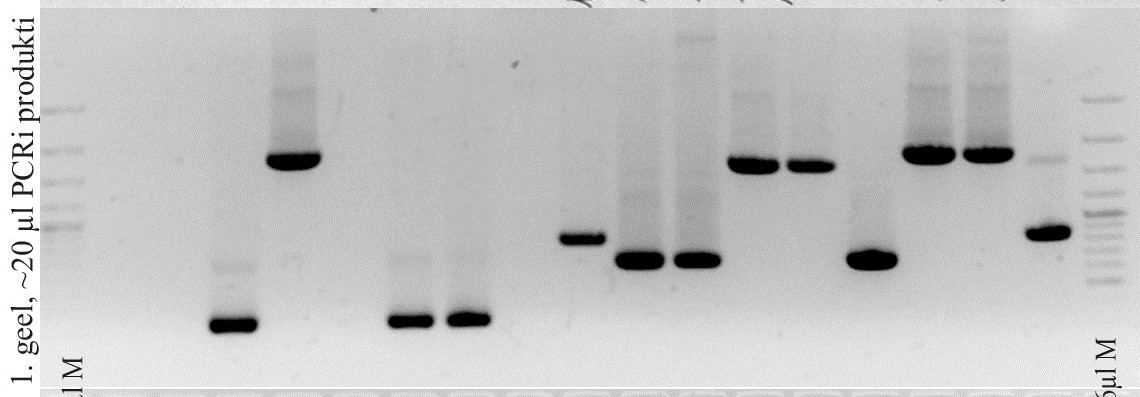
i. Tühja plasmiidi lõikamisel oleks bändide pikkused 17 bp (mida
on geelipildil praktiliselt võimatu näha, kui kontsentratsioon pole
just väga kõrge) ja 3566 bp. Kui insert on sisenenud, on tekkinud
bändide pikkused 360 bp ja 3566 bp. Pildilt on näha kahte bändi
kõrgemal, kui 3000 – üks on ilmselt õige pikkusega bänd (3566)
ja teine ilmselt terve plasmiid, mis on jäänud lõikamata (näiteks
seetõttu, et lõikamisaeg oli liiga lühike või tingimused polnud
optimaalsed). Lisaks on heledalt näha lõiku paarisaja bp kõrgusel
(kuna geel on üsna lühikest aega jooksnud, ja triibud on tihedalt
koos, on raske määrata täpset pikkust), sellest võib järeldada,
et insert on plasmiidi sisenenud.
ii. Pildi järgi on raske 100% kindel olla, kuid tundub, et bändid
on õige pikkusega.
7. a)
AATCTGAATTCGTCGACAAGCTTCTCGAGCCTAGGCTAGCTCTAGACCACACGTGTGGGGGCCCGAGCTCGCGGCCGCTGTATTCTATAGGGGAAAGCTCGGTACCACGCATGCTGCAGACGCGTTACGTATCGGATCCAGAATTCGTGATTTGAATTCGAGACCAACCTTTCCACCAAGATCTCCATCCTGGAACCCGAGACGGGGAAGGACATGTCCCCATACGAGGCCTACAAGAGGGGCATCATCGACAGGGGCCAGTACTTGCAGCTGCAGGAGCTCGAGTGTGACTGGGAGGAGGTCACCACCTCGGGGCCCTGTGGGGAGGAGTCTGTGCTCCTGGACCGCAAGAGCGGGAAGCAGTACTCCATCGAGGCCGCCCTCCGCTGCCGGCGCATCTCTAAGGAGGAGTACCATCTGTACAAGGACGGCCACCTGCCCATCTCCGAGTTTGCGCTGCTTGTAGCTGGGGAGACCAAGCCAAGCTCCGGATCCA
Tumedaga allajoonitud osa on
vektoris tagurpidi .
Sinisega märgitud A kohal on
toimunud mutatsioon , selle asemel on C. Selle tulemusel muutub ka
valgu järjestus.
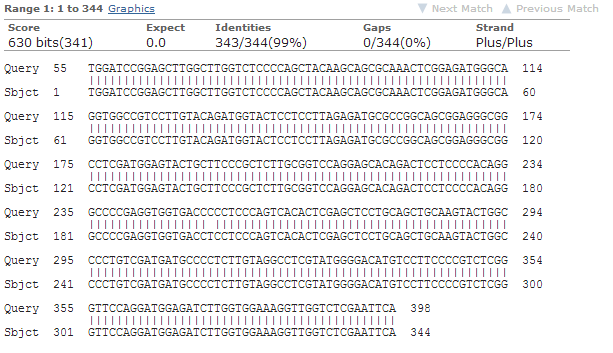
b) Sekveneerimise tulemuse järgi kloneerisin inimese envoplakin
geeni (Homo sapiens envoplakin (EVPL))
c) i. Saadud sekventsi pikkus oli 1049 bp. Sekventsi algus ning lõpp
olid kehvema kvaliteediga, keskmise osa kvaliteet oli küllaltki hea, piigid joonistusid selgelt välja ning olid loetavad.
ii. Sekveneeritud sai terve sisestatud insert.
iii. Järjestuses on tekkinud üks punktmutatsioon, kus A on
muutunud C-ks. See muudab ka aminohapet – E asemel sünteesitakse
A.
8.a) Kloneerimiseks tuleb vektorit ja inserti lõigata, tekitades
komplementaarsed kleepuvad otsad, seejärel tuleb otsad kokku
ligeerida.
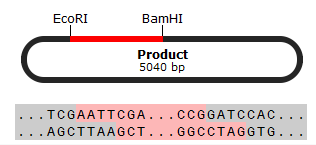
i. Restriktaasidest võiks kasutada EcoRI ja BamHI-d, et saada
vektorile ja inserdile sobivad „kleepuvad otsad”. Ainult
EcoRI-st ei piisa, kuna inserdil on vaid üks EcoRI lõikamissait –
ühte otsa tuleb töödelda mingi teise restriktaasiga, BamHI sobib
kõige paremini, kuna lõikab pärast kodeerivat järjestust. Kuna
inserdi algus ja lõpp on erineva restriktaasiga lõigatud, siis see
kindlustaks ka selle, et insert siseneb vektorisse õiget pidi.
Puhvritest oleks restriktsioonil kõige optimaalsem kasutada sama
firma poolt müügilolevat spetsiaalset EcoRI ja BamHI puhvrit,
ligeerimisel ligeerimispuhvrit.
ii. Jälgida tuleb, et lugemisraam ühtiks sisestatava valku
kodeeriva järjestuse lugemisraamiga. Minu kasutatud plasmiidile
(L-Envo pEGFP-C2) sobib pEGFP-C2, kuna Kui sisestataval järjestusel
pole stopp - koodonit , peab olema stopp- koodon kindlasti vektoris.
b) Skemaatiline esitus:
Vektor + insert:
9. a. Loomarakke säilitatakse pikemaajaliselt vedelas lämmastikus
külmutatuna, lühiajaliselt sobib ka -80oC. Külmutades
tuleb rakkudele kindlasti lisada dimetüül sulfoksiidi ja seerumit,
mis kaitsevad rakke. Rakud tuleb maha jahutada võimalikult
aeglaselt.
b. Rakkude sulatamine toimus peos soojendades. Sulatamine peab olema
võimalikult kiire, et rakud ei jõuaks DMSO keskkonnas soojeneda.
Pärast sulatamist tuli kiiresti lisada sooja (+37 oC)
söödet, et lahjendada DMSO lahust piisavalt, et see poleks
rakkudele enam toksiline . Kõige olulisem rakkude sulatamisel ongi
kiirus ning eelsoojendatud söötmete kasutamine, et rakud ei satuks stressi.
c. NIH3T3 rakud on pärit NIH Šveitsi hiire embrüost. Tegemist on
rakuliiniga, st rakud on võimelised piiramatu arv kordi paljunema.
Rakud sobivad kasvatamiseks kinnitunult plastikule või klaasile. Mikroskoobi all vaadates oli näha, et rakud paiknesid küllaltki
tihedalt üksteise vastas ning olid pikliku kujuga.
d. Rakke kasvatasime DMEM söötmes (sisaldab vett, soolasid,
süsivesikuid, vitamiine, aminohappeid, valke ja peptiide, rasvhappeid ja lipiide , mikroelemente ja pH indikaatorit), sellele
lisasime 10% veise loote seerumit (FBS), mis sisaldab samuti
erinevaid toitaineid, ning PEST-i. FBS on vajalik, kuna kunstlikult
valmistatud söötmes ei pruugi olla sobiv kogus vajalikke
toitaineid, lisaks on odavam lisada söötmele seerumit, kui kõiki
vajalikke toitaineid eraldi. PEST on antibiootikum, mis on vajalik
saastumisohu vähendamiseks.
10. a. Enne trüpsineerimist eemaldasin rakkudelt söötme ning
pesin rakke PBS-iga. Seejärel lisasin rakkudele trüpsiin-EDTA
lahust ning inkubeerisin 3 minutit. Lisasin söödet ning
suspendeerisin rakud plaadilt lahti.
i. PBS-iga pesemine on vajalik , et eemaldada söötmejäägid,
surnud rakud ja seerum , mis takistavad trüpsiini toimimist.
ii. Liiga kaua inkubeerides hakkab trüpsiin lagundama ka rakku
ennast, mitte ainult rakku plaadile siduvaid valke.
b. Rakkude tihedus plaadil oli umbes 50%, kuna plaadi suurus jäi
samaks, tuli 10% tiheduse saavutamiseks plaadile alles jätta 1/5
rakususpensioonist. Kokku oli 1200 μl suspensiooni, seega plaadile
jäi 240 μl ning eemaldasin 960 μl.
11. a. Lüüsipuhvri valmistamiseks pipeteerisin 15 ml tuubi
Tris-HCl (hoiab puhvri pH sobivana), NaCl, Triton-X 100 ( detergent ,
mis lõhub membraanstruktuurid, see vabastab valgud rakusisemusest ja
organellidest), glütserooli, EDTA (inhibeerib nukleaase, mis muidu
lagundaksid valgud) ning vett. Segasin ning hoidsin puhvrit jääl.
b. Sellist valgulüsaati kasutatakse tavaliselt edasi
immuunosadestamiseks, Western blotiks või ELISA analüüsiks. Need
meetodid võimaldavad uurida rakkude valgulist koostist või eraldada
spetsiifilist valku ülejäänud raku komponentidest.
12. a. Kasutasime keemilist meetodit. Selleks lahjendasime DMEM-is
eraldi DNA ja PEI ning seejärel segasime lahused kokku. DEI
moodustab DNA-ga kompleksid . Pipeteerisime tranfektsioonisegu
plaadile ning lisasime rakususpensiooni. DEI soodustab DNA seostumist
rakumembraaniga, fagotsütoosi teel sisenevad DNA:DEI kompleksid
rakku.
b. Tranfektsiooniprotokoll:
1. Rakkude vaatlemine mikroskoobis, rakkude seisukorra ja tiheduse
hindamine.
2. Transfektsioonisegu ettevalmistus. Kõigepealt tuleb lahjendada
seerumvabas rakusöötmes
(DMEM) eraldi epsides DNA ja PEI. Võta kolm 1,5 ml epsi. Pipeteeri
kõigepealt DMEM, siis lisa esimesse kahte epsi á 0,5 µg plasmiidi
(DNA) lahust (kontsentratsioone vaata lisa 2);
Kolmandasse epsi sega kokku kõigi transfektsioonide kohta DMEM ja
PEI (1µg DNA kohta 2µl PEI-d). DNA:PEI suhe sõltub rakuliinist,
vaja optimiseerida .
25 µl DMEM + pEGFP (250 ng/µl), 500 ng = 2 µl.
25 µl DMEM + pEGFP/FoxO3a (1000 ng/µl) 500 ng = 0,5 µl.
2x = 25 µl DMEM + 2x1 = 2 µl PEI lahust
3. Jagada DMEM+PEI segu võrdselt DNA segudele (26 µl kaupa), segada
läbi, tsentrifuugida ja inkubeerida 10-15 minutit toatemperatuuril.
Selles etapis tekivad DNA ja transfektsioonireagendi PEI kompleksid,
lahus muutub häguseks.
4. Tõsta katteklaasid steriilsete pintsettide abil 24-augulise
plaadi kahte auku.
5. Pipeteerida transfektsioonisegud klaasidele.
Aja kokkuhoiu mõttes jätsime meie ära tsentrifuugimise ning
pipeteerisime transfektsioonisegu kohe klaasile ning alles seejärel
inkubeerisime 15 minutit.
Rakkude külvamine 24-augulisele plaadile
6. Rakkude vaatlemine mikroskoobis. Rakkude tihedus umbes 100%
7. Rakkudelt imeda vaakumiga sööde, pesta 1 ml PBSiga , PBS ära
imeda.
8. Lisada trüpsiin-EDTA lahust 0,2 ml, inkubeerida mitte rohkem kui
5min.
9. Lisada 1ml söödet (DMEM + 10% FBS NB! antibiootikumideta),
suspendeeri rakud 1ml
automaatpipetiga tassi küljest lahti.
10. Teha söötmega sobiv lahjendus, et rakkude lõpptihedus
24-augulisel plaadil oleks 33,3%.
Arvestada 0,5 ml rakususpensiooni augu kohta. Selleks võtta 40 µl
rakususpensiooni ja 410 µl söödet augu kohta.
11. Pipeteeri rakususpensioon klaasidel olevatele
transfektsioonisegudele, loksuta.
12. Aseta rakud inkubaatorisse: 37°C, 5% CO2
13. 2-4h möödudes pesta rakke 1xPBS-ga ja lisada rakkudele värske
sööde (nüüd võib sisaldada ka antibiootikume).
Tavaliselt rakkude transfekteerimisel nad külvatakse, lastakse paraja tiheduseni (30-70%) kasvada ja transfektsioonisegud lisatakse
kinnitunud rakkudele. Meie kasutame vastupidist tehnikat aja
kokkuhoiu huvides.
c. Rakud elasid tranfektsiooni üle edukalt – mikroskoobi all oli
näha, et rakkude tihedus oli kasvanud uuesti 100%-ni.
d. Transfektsiooni efektiivsus oli umbes 30%.
13. a. Rakkude fikseerimise ja antikehadega märgistamise protokoll:
Vajalike lahuste valmistamine:
1. Fikseerimislahus: 4% PFA 1x PBSis, 10ml.
a. 0,4g PFA puru kaaluda väiksesse keeduklaasi
b. lisada 1ml 10x PBSi ja 9ml vett
c. soojendada magnetsegajal tõmbe all kuni puru on lahustunud.
2. 1xPBS, 50ml.
a. 5ml 10x PBS-i.
b. Lisada sellele vett (MQ) 50 ml-ni, segada.
3. PBS- Tween : 1xPBS, millele on lisatud 0,05% Tween 20, 50ml
a. 50ml 1xPBS-ile lisada 50 µl 50% Tween-20 lahust, segada.
4. Permeabiliseerimislahus: 50mM NH4Cl , 0,5% Triton X-100 PBSis, 5ml
a. 0,25 ml 1M NH4Cl varulahust
b. 0,25 ml 10% Triton X-100
c. 0,5 ml 10x PBS-i
d. Lisada vett 5 ml-ni, segada (mitte vahtu ajada).
5. Blokklahus: 2% BSA, 0,1% NaN3 PBSis, 2ml
a. 0,04 g BSA-d
b. 0,02 ml 10% NaN3
c. Lisada 1xPBS-i 2ml-ni.
I Fikseerimine
1. Rakkudelt eemaldada vaakumiga sööde
2. Rakud fikseerida 0,5 ml 4% PFAga PBSis, inkubeerida 15 minutit
toatemperatuuril.
3. Pesta rakke 3 korda 5 minutit á 1 ml 1x PBSiga.
II Neutraliseerimine ja permeabiliseerimine
1. Lisada rakkudele á 0,5 ml 50mM NH4Cl + 0,5% Triton X-100 PBSis,
inkubeerida 15 minutit toatemperatuuril.
2. Pesta rakke 2 korda 5 minutit á 1 ml 1x PBSiga.
III Blokeerimine
Lisada rakkudele á 0,5 ml 2% BSA PBSis, millele on lisatud 0,1%
NaN3, jätta seisma üleöö (üle nädalavahetuse) +4°C juurde.
IV Inkubatsioon primaarse antikehaga
1. Valmistada primaarse antikeha lahjendused (kulub 200 µl ühe
klaasi kohta) 0,2% BSA-PBS lahuses
a. 40 µl 2% BSA lahust
b. 360 µl 1x PBSi
c. Mouse anti tubuliin 1:500 0,8 µl, segada.
2. Lisada rakkudele ja inkubeerida 60-90 minutit toatemperatuuril.
3. Pesta 3 korda 5 minutit á 1 ml PBS-Tweeniga.
V Inkubatsioon sekundaarse antikehaga.
1. Valmistada sekundaarse antikeha lahjendused 0,2% BSA-PBS lahuses
(vaja on kahe klaasi
jagu, seega kokku 2x200=400µl).
a. 40 µl 2% BSA lahust
b. 360 µl 1x PBSi
c. Goat anti mouse Alexa546 1:2000 0,2 µl, segada.
2. Inkubeerida 45-60 minutit toatemperatuuril, kaitsta valguse eest.
3. Pesta 3 korda 5 minutit 1 ml PBS-Tweeniga.
4. Pesta 2 korda 5 minutit 1 ml PBSiga.
5. Pesta 1 kord 1 ml milliQ (destilleeritud) veega.
VI Alusklaasidele panemine ehk sulundamine
1. Kirjuta alusklaasile oma nimi ja kuupäev, samuti kasutatud
antikeha.
2. Pigistada tuubist alusklaasidele väike tilk DAPIga
sulundamislahust.
3. Võta katteklaas pintsettide vahele ja kuivata serv vastu pehmet
paberit.
4. Aseta katteklaas (rakud allpool !!!) sulundamislahuse tilga sisse
(väldi mulle).
5. Lasta kuivada ja säilitada pimedas .
b. Lahjenduste tegemise arvutused:
1) Primaarse antikeha lahjendus: Vaja on 400 µl lahust, kuna meil on
2 klaasi, kummagi kohta 200 µl. 2% BSA tuleb lahjendada 0,2%, seega
tuleb lahjendada 10x, 10% lahusest peab olema BSA, seega tuleb võtta
400/10=40 µl. Antikeha peab olema 1:500, seega tuleb lahjendada 500
korda, seega 400/500=0,8 µl.
2) Sekundaarse antikeha lahjendus: BSA lahjendus sama, mis primaarse
antikeha puhul. Antikeha peab olema 1:2000, seega tuleb lahjendada
2000 korda, 400/2000=0,2 µl.
c. Fikseerimine on vajalik, et kinnitada rakud plaadile ning
fikseerida antigeenide asukoht rakus, samal ajal säilitades raku
natiivne kuju. Neutraliseerimine on vajalik selleks, et fikseerimisel
kasutatud ained ei segaks antikehade seondumist ning
permeabiliseerimine võimaldab antikehadel rakku siseneda ning
pääseda antigeenideni. Blokeerimise eesmärk on takistada
antikehade ebaspetsiifilist seondumist nendesse kohtadesse , kuhu
igasugused valgud kleepuvad.
Primaarse antikehaga inkubeerimisel seondub primaarne antikeha
antigeeniga – antud juhul tubuliiniga. Inkubeerides sekundaarse
antikehaga seonduvad sekundaarsed antikehad primaarse antikehaga.
Kahekihiline reaktsioon on vajalik selleks, et poleks vaja iga primaarset antikeha fluorokroomiga konjugeerida, lisaks võimendab
see signaali, kuna iga primaarse antikehaga saab seostuda mitu
sekundaarset antikeha.
Alusklaasidele panemine on vajalik mikroskopeerimiseks.
Sulundamislahus sisaldab DAPI -t, mis seondub DNA-ga ning
fluorestseerub siniselt , see võimaldab fluorestsentsmikroskoobiga
näha ka DNA-d.
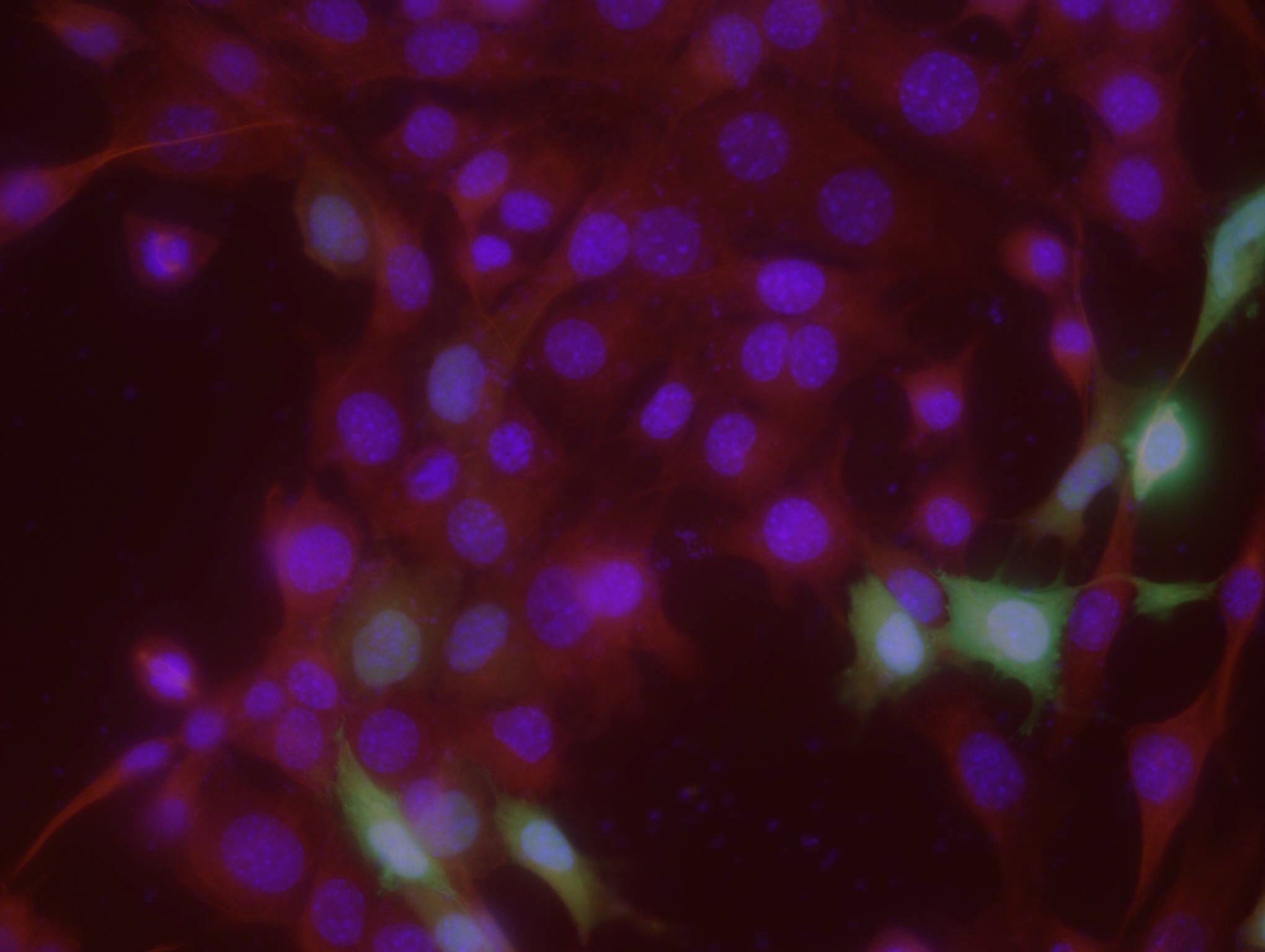
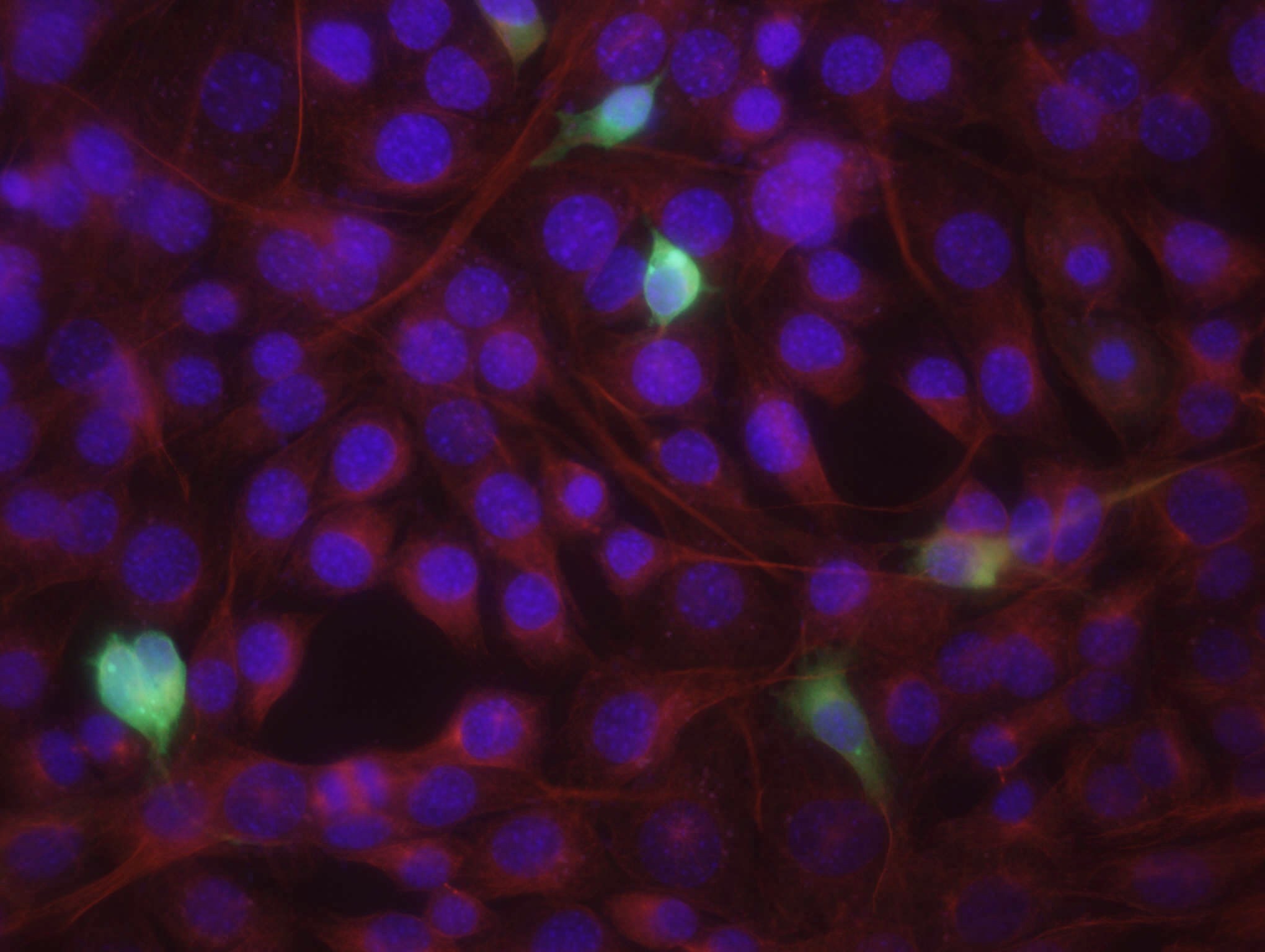
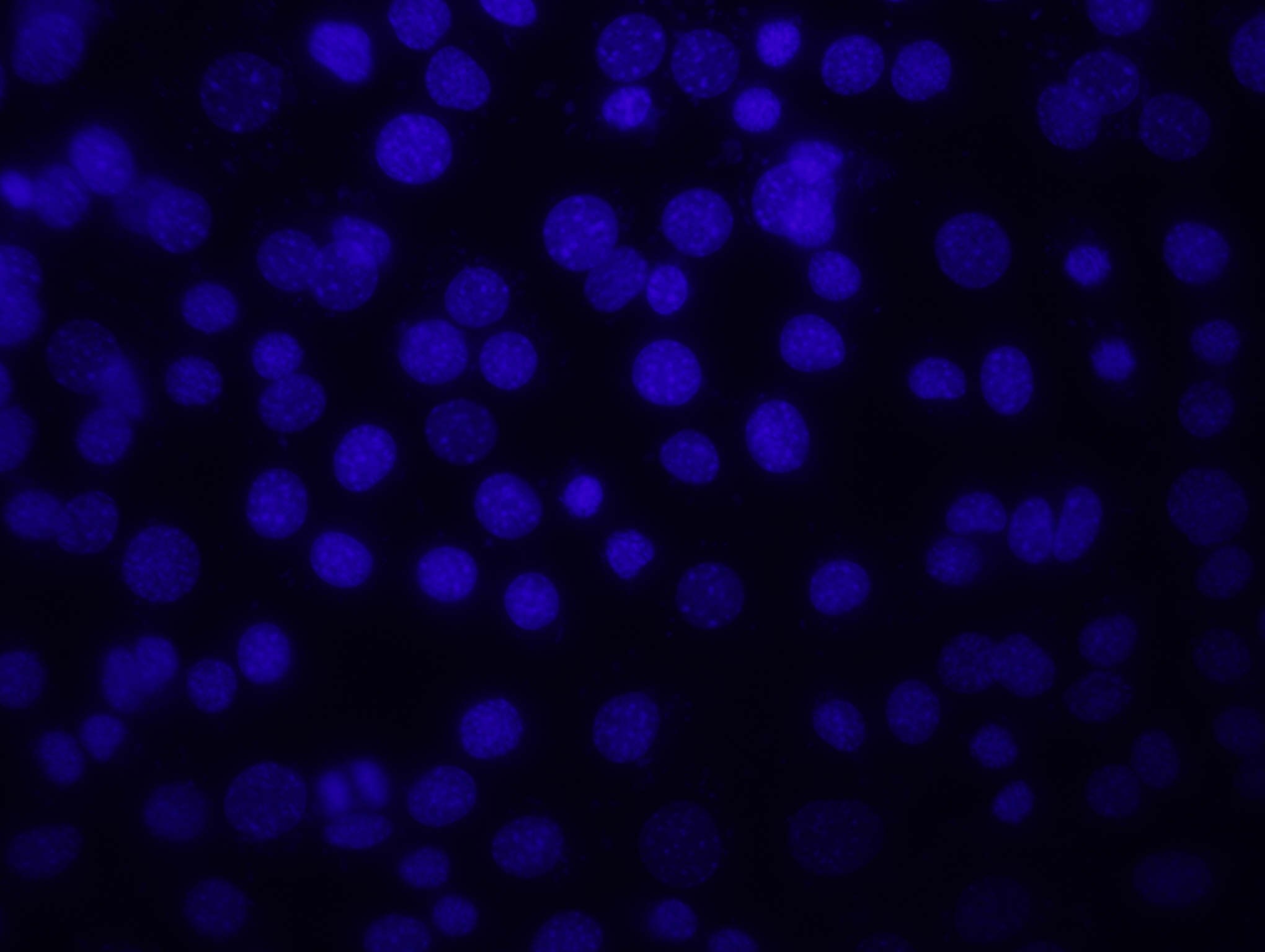
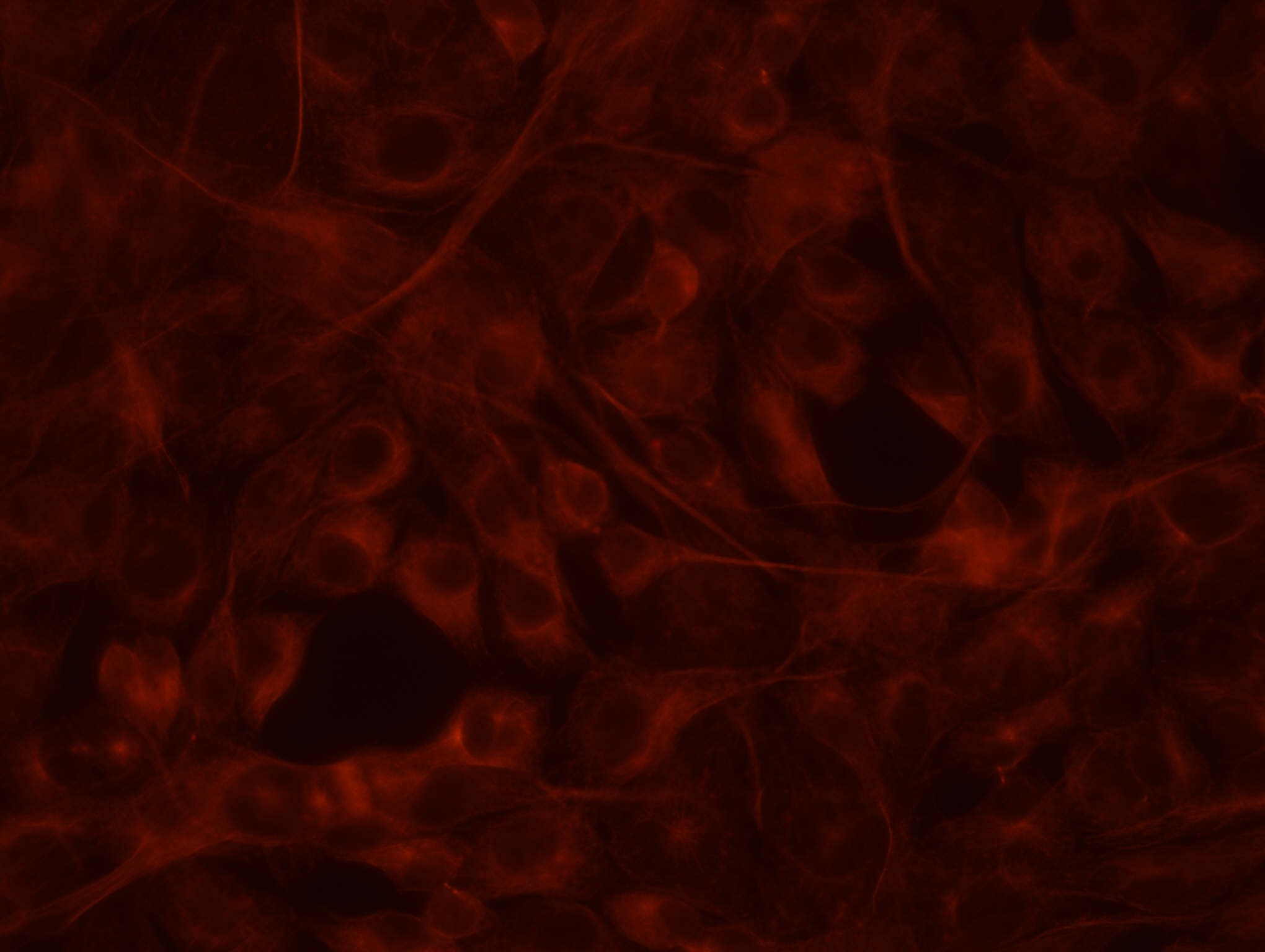
14. a. Kontroll-plasmiidiga rakud:
Pildil on näha siniselt DNA, roheliselt edukalt transfekteeritud
rakkudes ekspresseeritud GFP ning punaselt antikehaga värvitud
tubuliin.
pEGFP FoxO3a-ga transfekteeritud rakud:
b. Valke detekteerisin kahekihilise reaktsioonina. Primaarseks
antikehaks oli Mouse anti tubuliin 1:500 ja sekundaarseks antikehaks
Goat anti mouse Alexa546 1:2000.
c. Sekundaarne antikeha on valitud kasutatud primaarse antikeha
järgi. Kuna primaarne antikeha oli tehtud hiires, kasutasime kitses
toodetud antikehasid, mis tunnevad ära hiire primaarsed antikehad
(mouse anti tubuliini). Need on konjugeeritud punase Alexa546
fluorokroomiga.
d. Kasutasime sinist, punast ja rohelist fluorokroomi. Sinine oli
DNA värv DAPI, mille ergastamiseks on vaja lainepikkust ~360 nm, kui
värv on seostunud kahe-ahelalise DNA-ga, ja 341 nm (jääb UV
kiirguse ja lilla valguse piiri peale), kui värv pole DNA-ga
seostunud, ning kiirgab valgust lainepikkusega 450-460 nm. Punane oli
sekundaarse antikehaga kojungeeritud fluorokroom Alexa546, mille
ergastamiseks on vaja valgust lainepikkusega 546 nm (roheline valgus)
ning kiirgab valgust lainepikkusega ~600, mis on oranžikas-punast
värvi.
Roheline oli EGFP – enhanced green fluorescent protein , mida kodeeriv järjestus sisaldus kasutatud vektoris. Ergastamiseks on
vajalik valgus lainepikkusega ~488 nm (sinine valgus) ning
emiteeritava valguse lainepikkus on ~520 nm, mis on roheline.
e. Mõlemal pildil on näha punasega ja sinisega kõik plaadil olevad
rakud ning rohelisega edukalt transfekteerunud rakud. Arvestades kogu
rakkude ja transfekteerunud rakkude suhet, saab hinnata
transfektsiooni edukust , mis on umbes 7% FoxO3a-ga ja 14%
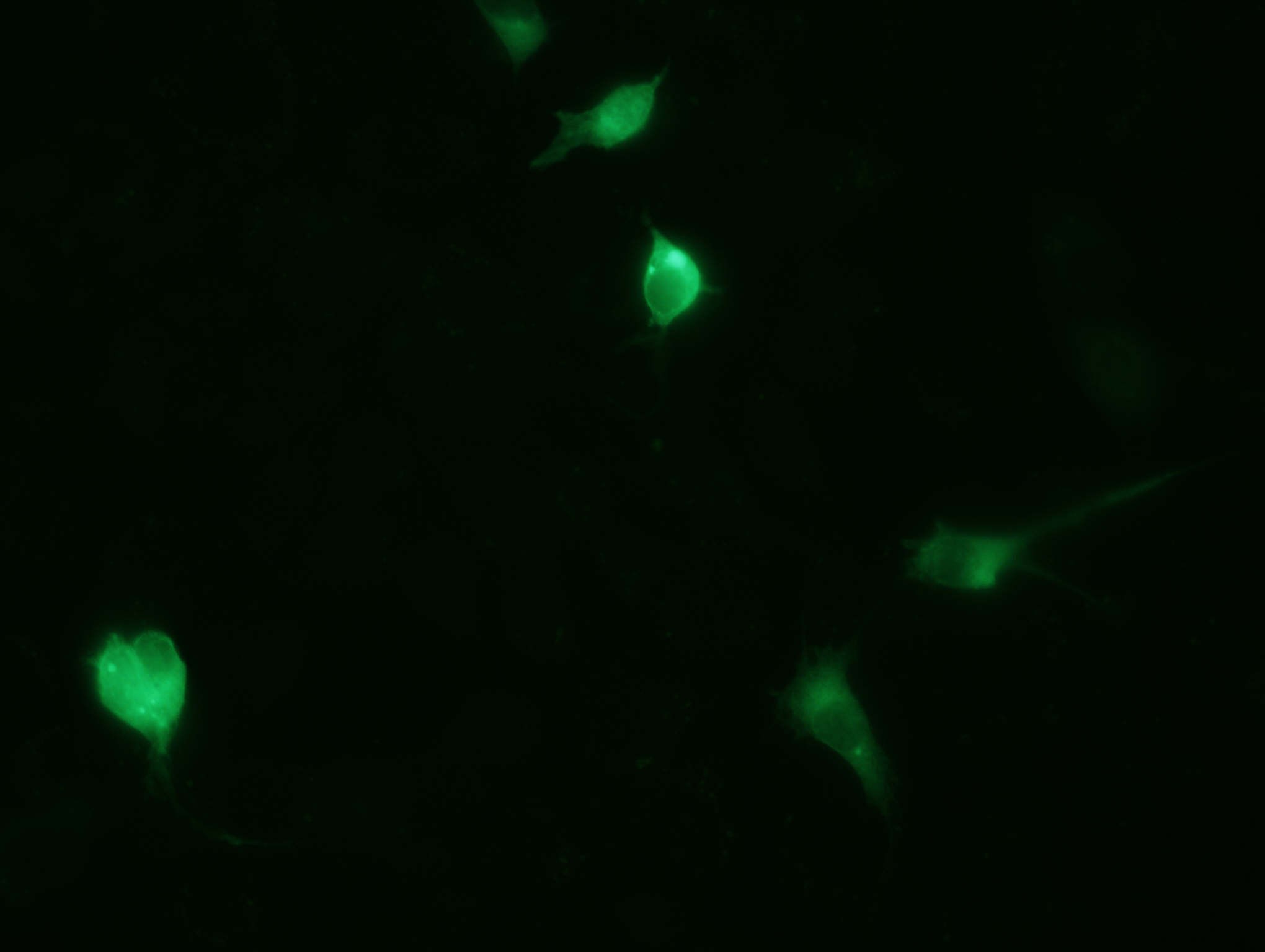
kontrollplasmiidiga. Allolevalt pildilt on näha, et osad rakud
ekspresseerivad GFP-d väga suures koguses (seda leidub nii tuumas
kui ka tsütoplasmas), osad aga väiksemas koguses (seda leidub
ainult tsütoplasmas), osad rakud on vaevumärgatavalt heledad, seega
GFP kogus on seal väga väike. Enamikus kontrollplasmiidiga rakkudes
leidub GFP-d nii tsütoplasmas kui ka tuumas, samas FoxO3a-ga
rakkudes on GFP peamiselt tsütoplasmas. See on selgitatav sellega,
et FoxO3a paikneb tavaliselt tsütosoolis, seega FoxO3a-ga koos
ekspresseeritud GFP paikneb samuti tsütosoolis. Teatud
ellujäämisfaktorite puudumisel translokeerub FoxO3a tuuma, mis
selgitab seda, miks osades rakkudes leidub GFP-d nii tsütoplasmas
kui ka tuumas.
Sinise värvi järgi saab leida rakkude koguarvu, kuna värvunud on
DNA. Pildilt on näha, et tuumad on täpilised – see on põhjustatud
DNA ebaühtlasest paiknemisest rakus.
Punaselt on värvunud tsütoplasmavõrgustik, näha on värvumata
ümarad tuumad.
Rakkudest lüsaadi valmistamine
BCA töölahuse valmistamine
Standardvalguga kalibreerimiskõvera jaoks valmistasime 1000 µl
lahust, oma proovi jaoks 440 µl lahust (400 µl + 40 µl varu). Oma
lahuse jaoks võtsin 431,4 µl reagenti A ja 8,6 µl reagenti B.
Segasin hoolega ja jagasin 100 µl kaupa 96-se plaadi aukudesse.
BSA lahjendusterea ettevalmistus
Võta 5 epsi, kirjuta peale kontsentratsioonid
Pipeteeri igasse 50 µl lüüsipuhvrit
Võta 50 µl BSA 2 mg/ml lahust ja lisa esimesse epsi, suspendeeri korralikult.
Tõsta sama otsikuga 50 µl lahust järgmisse epsi ja suspendeeri.
Korda tegevust, kuni oled jõudnud 0,125 mg/ml epsini.
Viimasesse epsi jääb puhas puhver , ära sinna midagi juurde pane.
Oma proovi ettevalmistus
Tsentrifuugisin valguprepsi 1 minut max pööretel. Tegin uude epsi
2x lahjenduse – 15 µl lüsaati ja 15 µl puhvrit.
Reaktsiooni kokkusegamine
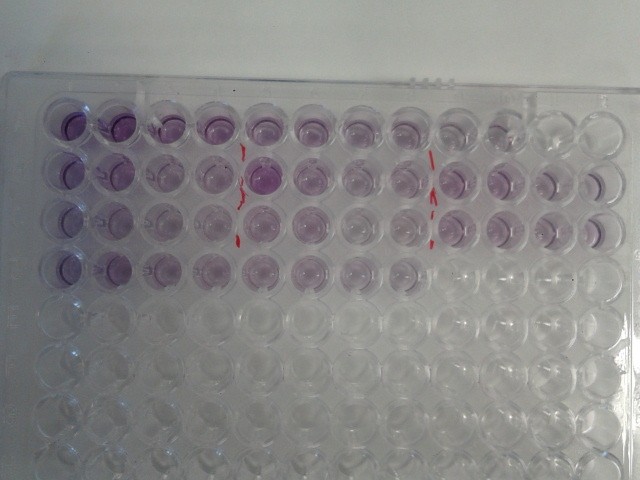
Lisa iga proovi 5 µl kahte auku. Raputa plaati segamiseks, aurumise vältimiseks kata plaat teibi või kaanega. Inkubeeri 37 kraadi
juures 30 minutit. Mõõda neeldumist 562 nm juures.
Meie proovid on näha B-reas 1-4.
y = 0,2601x + 0,1567
Kontsentreeritud:
0,2502
0, 2345
2x lahjendus:
0,1833
0,19
Valgu kontsentratsioonid:
0,359
0,299
0,205
0,333
Keskmine: 0,299 mg/ml







































Kõik kommentaarid