Molekulaar -
ja rakubioloogia
praktikum ARUANNEAve
Tüür
155356
YAGB41
Kevadsemester
2017
Pipeteerimine
8.
veebruarI HARJUTUS
Eesmärk:
Õppida õigesti pipeteerima.
Materjalid: Arvutused:
- Pesuvahendi kontsentraat Pesuvahendit on vaja:
- Vesi = 1,65ml
- Pipetid ja otsikud
- 15ml falkon tuub
- 1,5 ml tuubid
- PCR plaadi tükk
Töö käik: - Teen 15-ml falkon tuubi 5,5ml 30% homogeenset pesuvahendilahust
- Võtan kaks 1,5ml- tuubi ning pipeteerin mõlemasse 1,55ml lahust.
- Pipeteerin alles jäänud lahuse 200µl kaupa PCR plaadi aukudesse.
Tulemus:
Sain 7,5
auku täidetud, kuigi oleks pidanud jätkuma 12 tuubi jaoks.
Viga tuli sellest, et detergenti pipeteerides jäid sisse õhumullid,
mida tegelikkuses ei oleks tohtinud seal olla.
II HARJUTUS
Eesmärk:
Õppida silma järgi
hindama pipeteeritava koguse mahtu.
Materjalid:
- Erinevate veekogustega tuubulid
Töö käik: - Esimeses tuubulis on 200µl
- Hinnata silmaga, mis kogused on järgnevates tuubulites.
Tulemused:
Minu arvatud
Tegelik
200 µl
170µl
150µl
120µl
100µl
80µl
500µl
130µl
120µl
20µl
10µl
60µl
60µl
50µl
20µl
Hindaksin oma
täpsust rahuldavaga. Kindlasti kogemusega see
paraneb .
III HARJUTUS
Eesmärk:
Harjutada iga pipeteerimiskorraga sama mahtu pipeteerima.
Materjalid:
- Broomfenoolsinine
- Äädikhappe lahus
- Blotipaber
- Pipetid
- PCR plaat
Töö käik: - Pipeteerin plaadi nelja auku 100µl broomfenoolsinist
- Pipeteerin lisaks vastavalt 50µl; 5µl; 0,5µl äädikhappe lahust.
- Pipeteerin blotipaberile oma nime ja harjutuseks täpikesi
Tulemus:
Tulemus ei olnud väga
imeline , kuna algselt ma ei
pannud tervet pipetitäit üheks täpiks vaid jagasin selle kaheks. See on aga
harjutuse eesmärgiga
vastuolus . Kohe kui oma veast aru sain hakkasid
ka ilusamad ja ühtlasemad
täpid tulema .
Praktiline töö nr.
1: Polümeraasi ahelreaktsioon
Eesmärk:
Paljundada valitud valku või valgu mõnda domääni kodeerivat DNA
järjestust ehk inserti PCR meetodiga.
Materjalid:11,3 µl
MQ eriti puhas vesi
lahusti
2,0 µl
10x polümeraasi puhvrit
loob sobiva keskkonna, et
polümeraas töötaks
2,0µl
25mM
MgCl2 (lõppkontsentratsioon 2,5mM)
on polümeraasi ko-faktoriks, seega mõjutab reaktsiooni spetsiifilisust
2,4µl
2mM dNTP (lõppkontsentratsioon 240µM)
et segus oleks vabu
nukleotiide 1,0µl
Pärisuunalist praimerit (lõppkontsentratsioon 0,5µM)
Polümeraasi seondumiseks 5´ poolele
1,0µl
Vastassuunalist praimerit (lõppkontsentratsioon 0,5µM)
Polümeraasi seondumisesk 3´ poolele
0,2µl
template-DNA (lõppkontsentratsioon 0,25ng/µl)
sisaldab regiooni, mida tahetakse praimerite abil amplifitseerida
0,1µl
HotFIRE Pol Polümeraas (0,5U)
Termostabiilne polümeraas, sünteesib 5´-3´
sidemeid .
Arvutused: Kokku
soovime saada 20µl
Polümeraas: Magneesiumkloriid: = 2µl
Praimerid: dNTP:
Template DNA
lõppkontsentratsioon:
Polümeraasi
ühikud :
Vesi: 20 – 2 –
2 – 1 – 1 – 2,4 – 0,2 – 0,1 = 11,3µl
Töö käik:
- Segan kokku eelnimetatud komponendid
- Tsentrifuugin ja asetan PCR masinasse
- PCR programm:
- 95 kraadi juures 15 minutit – polümeraasi aktiveerimine
- 95 kraadi juures 10 sekundit – DNA denatureerimine
- 56 kraadi juures 20 sekundit praimerite hübridiseerimine
- 72 kraadi juures 1 minut – ekstensioon
- Korrata alates punktist b. 24 korda.
- 10 minutit 72 kraadi juures, et pikendada poolikuks jäänud PCRi produktid
Millist
template DNA-d ja praimereid kasutasin ? Mille järgi
valitakse PCR praimerite hübridiseerimise ehk annealing temperatuur?
Milline sulamistemperatuur on praimeril l 5’- GCCATCTCATGCCCATCGC -3’ standardtingimustel ja millist hübridiseerimistemperatuuri
kasutaksin? Kuidas need leidsin?Hübridiseerimise temperatuur ja aeg sõltub kasutavate praimerite
sulamistemperatuurist, amplifitseeritava DNA konsentratsioonist ja
reaktsioonisegu koostisest. Antud järjestuse sulamistemperatuuriks
on 59,3 kraadi, mille leidsin:
http://eu.idtdna.com/site/Order/oligoentry/set?seq=GCCATCTCATGCCCATCGC .
Kasutaksin hübridiseerimistemperatuuriks 59 kraadi.
Mille järgi
valitakse ekstensiooniaeg? Kui pikk see oleks 2000bp produkti korral,
kasutades samu reagente nagu praktikumis ?
Ekstensiooniaeg
sõltub amplifitseeriva järjestuse pikkusest ja G/C-nukleotiidide
osakaalust. Kuna meie poolt kasutatud HotFIREPol sünteesib
keskmiselt 1kb DNAd ühe minuti jooksul, siis läheks teoreetiliselt
sellel aega 2 minutit, aga anname natukene lisaaega ka ja ütleme et
2,5 minutit.
Kui esialgses
reaktsioonis on 4 molekuli matriits DNA’d, siis mitu molekuli PCRi
produkti on tekkinud pärast 25.tsükli lõppemist.
Tehniliselt võiks
mõelda et kui on 4 molekuli, siis see on 22. Ja tehtud on
2 ringi PCRi. Liidame selle 25le juurde ja saame tehniliselt 27
ringi, kui oleksime alustanud 1
molekuliga . 227=134217728
Praktiline töö nr.
2: Agaroosgeeli valamine
Eesmärk:
PCR produkti
analüüsimiseks vajaliku agaroosgeeli valmistamine.
Materjalid:
4,5g
Agaroosi pulber
Varieerides geeli agaroosisisaldust saame lahutada ka suhteliselt sarnaste pikkustega DNA fragmente.
300ml
1x TAE
puhver lahusti
3µl
EtBr (lõppkontsentratsioon 0,05µg/ml)
DNA visualiseerimise reagent
Arvutused:
Agaroosi pulber: EtBr: , aga meie
paneme EtBr 2x rohkem, kuna õppejõud ütles nii. Parem
karta kui kahetseda.
Töö käik:
- Lahustame agaroosi pulbri TAE puhvris
- Kuumutame seda mikrolaineahjus, et kõik pulber lahustuks
- Jahutame ja seejärel lisame EtBr.
- Kanname segu geelialusele, kuhu on eelnevalt lisatud hammaste tekkimiseks „kamm“
- Jälgida, et mulle ei tekiks ja eemaldada need.
Millise %
geeli valasime? Miks?Vaatasime lk 32
olemat tabelit nr. 6 ja otsustasime teha 1,5% geeli. Tabel näitab,
et see
geel sobib DNA’le
pikkuses 0,2-3kb ja see on igati sobilik
kõigi
grupis olevate inimeste DNA lahutamiseks, kuna meie DNA’de
pikkused jäid vahemikku 0,3-1,6kb.
Mis juhtub kui unustame EtBr lisada? Kas olukorda saaks parandada?
Kui me ei lisaks
EtBr, siis me ei näeks enda DNA bande pärast geelis. Olukorra
parandamiseks võib geeli EtBr’is solgutada. Oluline on, et geel
oleks
homogeenne ning et EtBr oleks ühtlaselt geelis jaotunud.
Kui oleks vaja
valada 0,75% geel 80ml TAE puhvrist, mitu g agaroosi oleks tarvis
võtta? Kui palju tuleks lisada EtBr kui alg- ja lõppkontsentratsioon
on samad praktikumis kasutatutega?Lahusti: 80 ml
Agaroosi on vaja:
80 x 0,75/100 = 0,6g
EtBr
algkontsentratsioon : 10mg/ml=10µg/µl EtBr lõppkontsentratsioon:
0,05µg/ml
Saame teada, mitu
µg meil on EtBr vaja: 80 x 0,05 = 4µg
Mitu µl on EtBr
vaja? 10/4 = 1/x ehk x = 4/10 = 0,4µl
Praktiline töö nr.
3: DNA lahutamine agaroosgeelis
Eesmärk:
PCR produkti lahutamine geelelektroforeetiliselt.
Materjalid:
18 µl
PCR
produkt DNA, mida visualiseerima
3,6 µl
DNA laadimispuhver
DNA visualiseerimiseks ja paremini hambasse vajutamiseks.
TAE agaroosgeel
GeneRuler 100bp Plus Ladder
Marker DNA
fragmentide pikkuse ning koguae ligikaudseks kvantifitseerimiseks
Arvutused:
Meil on 20 µl
PCR produkti, kust võeti 2 µl ära.
DNA
laadimispuhvri koguse jaoks teeme
arvutuse : 18/5 =3,6 µl
Töö käik: - Pipeteerin PCR segule 6x DNA laadimispuhvri.
- Pipeteerime markeri geeli esimesse hambasse.
- Pipeteerin enda segu geeli hambasse.
- Lahutame DNA geelis elektrivoolu toimel (150V ja laseme joosta 20 minutit)
- Teeme UV valguse all IQ400 geelipildistajas oma geelist pildi.
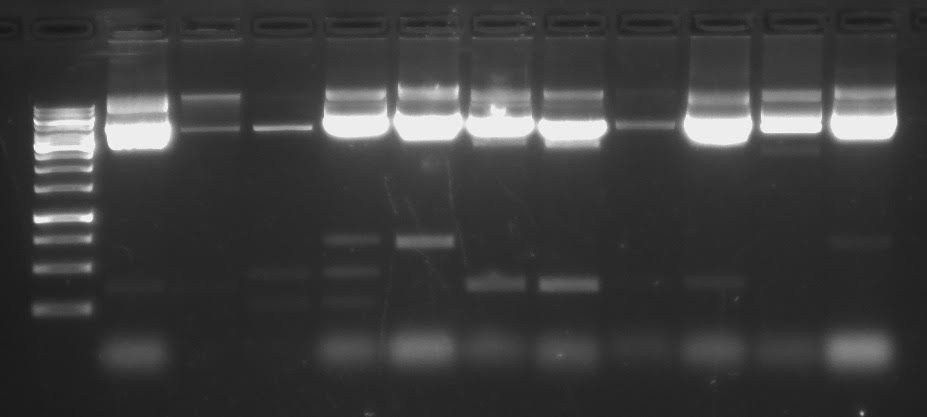
Tulemused:Minu rada: 11
Mida näen
geelis oma rajal? Mida saan sellest geelipildist järeldada?Näen oma
rajalt ,
et minu DNA
fragment on kuskil 600-700
aluspaari pikkune , mis on ka
täiesti
korrektne , kuna kasutasin Dystonini(DST), mis on eelantud
tabeli järgi 644 aluspaari pikkune. Samas saan järeldada, et DNA
kontsentratsioon on suur.
Mis juhtub,
kui DNA laadimispuvrit lisada arvutatust 2x rohkem? Mis juhtub, kui
seda PCRi produktile enne geelile pipeteerimist üldse ei lisata?Ei tohiks nagu
midagi juhtuda. Kui me puhvrit üldse ei lisa, siis ei näe pärast
oma DNA bände ja seda on algselt raksem hambasse pipeteerida. DNA
võib hambast välja voolata.
Mis juhtub
migreeruva DNA-ga, kui elektroforees „ununeb“ jooksma mitmeks
tunniks? Kus on DNA?
Mida kauem
hoidad, seda kaugemale DNA liigub. Lõpuks meil ei ole geelis enam
DNAd näha, sest DNA läheb puhvrisse.
Praktiline töö
nr.4: DNA puhastamine agaroosgeelist
Eesmärk:
Huvipakkuva PCR produkti väljapuhastamine geelist.
Materjalid:Võimalikult täpselt välja lõigatud (0,15g)
Agaroosgeeli tükk DNA fragmendiga
Meile huvipakkuv DNA fragment
450 µl
ADB puhver
Kõrge soolakonsentratrsioon ja sobilik pH, et DNA seonduks SiO2’le.
2 x 200 µl
DNA pesupuhver
Et kõrvalistest ainetest lõplikult
vabaneda .
10 µl
Elueerimispuhver
Madala soolasisaldusega aluseline solvent, et DNA rehüdreeriga ning räni küljest vabastada.
Arvutused: 0,1g
geeli kohta 300µl. 1,5x300=450µl
Töö käik:
- Lõikan skalpelliga geelist enda fragmendi välja. (0,15g)
- Lisan ADB puhvri geelile.
- Inkubeerime 55kraadi juures 5minutit, kuni geel on täielikult sulanud.
- Pipeteerin kogu koguse ränikolonni.
- Tsentrifuugin – DNA jääb räni külge ja ülejäänud puhvri viskan ära.
- Pipeteerin peale 200 µl pesupuhvrit, tsentrifuugin ja kordan.
- Tõstan kolonni puhtasse 1,5ml tuubi. Elueerin DNA kolonnist elueerimispuhvri abil. Inkubeerin paar minutit laua peal
- Tsentrifuugin 1 minuti. Seejärel on tuubi põhjas elueerinud PCRi produkt lahustatune elueerimispuhvris.
- Kontrollimiseks segan kokku 2 µl eluaati, 6,33 µl MQ ja 1,66 µl 6x värvi. Tsentrifuugin ja kannan geelile. Geeli pilt tulemuste all.
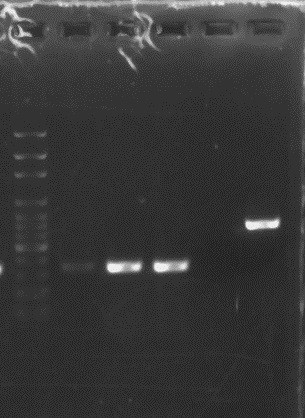
Tulemus:
Minu rada nr8.
Miks on vaja PCR produkti puhastada ?Tahame vabaneda kõigest üleliigsest lahuses, nätieks praimeritest,
nukleotiididest ja erinevatest sooladest, et need ei mõjutaks meie
järgmiseid
etappe .
Milleks on vaja teha kontrollgeel?
Esiteks, et vaadata, kas meie segus on ainult meid huvitav DNA
fragment. Teiseks, et kontrollida, kas elueerimisprotsess õnnestus,
kas
saime ikka kogu DNA ilusti ränist kätte ja tuli meile topsi?
Mida tänasest kontrollgeeli pildist järeldad ?Järeldan, et minu lahuses on puhtalt ainult
vajaminev DNA fragment,
ja et seda on väga palju.
Milline aine ja kuidas seob kolonnis DNA-d? Milline
elueerimispuhvri omadus aitab DNA kolonnist välja elueerida?Kolonnis olev ränioksiin seob DNA, nii, et DNA seob
fosfaatselgroogupidi ränioksiidiga. (seondumisele aitab kaasa,
eelnev ADB puhver). Elueerimispuhvri omadustest on kõige olulisemad
madal
soolasisaldus ja aluseline pH.
Millist olulist infot saan kolonnide kohta firma kodulehelt? Kas
kasutatud kit sobis antud praktikumi töö teostamiseks, millest seda
järeldada?Aeg
15 minutit
Vajatav
varustus Tsentrifuug
DNA suurus
50bp kuni 23kb
Sobivad
proovid DNA TAE/TBE puhvriga agaroosgeelist
DNA
puhtus Kõrge kvaliteedi, puhtusega DNA, mis on sobilik sekveneerimiseks, ligatsioonireaktsiooniks jne...
Kui palju DNA saadakse
50bp-10kb puhul 70-90%
Praktiline töö nr. 5: Rekombinantse plasmiidi
ligeerimine
Eesmärk:
Meid huvitava aplifitseeritud DNA järjestuse
sisestamine vahevektorisse ehk paljundusplasmiidi.
Materjalid:1,25 µl
pSTBlue-1
vektor (16ng/µl)
Lineaarne vahevektor, kus on
origin , antibiootikumide resistentsusgeen, lacZ üksus ja AccepTor kloneerimissait.
3,75 µl
PCR produkt
Sisaldab inserti
1 µl
10X ligeerimispuhvrit
Loob sobiva keskkonna
0,5 µl
T4 DNA
ligaasi (5U/µl)
Ligeerib inserdi ja vektori kokku
3,5 µl
MQ
lahusti
Arvutused:
Vektor: 16/1 = 20/x x = 1,25 µl
PCR produkt: 150ng/10µl=15ng/µl PCR vektrori suhe peab olema 3:1.
Ehk PCR võtame 3*1,25*3,7 µl
10x puhver: 10/10
= 1
Ligaas : 5U/ µl
tahame saada 2,5U ehk võtame 0,5 µl
MQ:
10-1,25-3,75-1-0,5 = 3,5
Töö käik: - Pipeteerin kokku ligeerimissegu
- Jälgin et lahus saaks homogeenseks
- Tsentrifuugin
- Jätan üleöö ligeerima +4 kraadi juurde.
Millise
konstrukti oma töö käigus kloneerisin?Ligeerisin
sellise konstrukti, kus bSTBlue-1 vektoris
EcoRV saidis on minu
insert ehk DST geen.
Milleks
kasutasime kloneerimisel pSTBlue-1 vektorit ? Mis teeb selle eriliseks ?See on meie
vahevektor ja see on väga hea kuna võimaldab sini-valge skriinimist
ja seal on mõlemal pool EcoRV saiti
EcoRI sait, ehk seda oli väga
lihtne restriktaasidega lõigata.
Kas sini-valge
selektsiooni kasutamisel võib õige koloonia olla ka sinine? Miks?
Vahel ikka
juhtub. Insert võib olla väga väike, seega lacZ jääb terveks ja
koloonia värvub ikkagi siniseks.
Kui tahame 5kb
suurusesse vektorisse ligeerida 1000bp pikkust inserti, siis kui
palju vektorit peame 50ng inserdi kohta võtma? Soovitud
vektori:inserdi suhe on 1:5.
Vektor : 5000bp Insert: 1000bp
10ng Kuna suhe
peab olema 1:5 50ng
Vektor on aga 5
korda pikem kui insert, seega on see ka 5x raskem massi poolest, ehk
peame selle 10ng viiega korrutama, et saada reaalse massi. Peame
võtma 50ng vektorit.
Eesmärk:
Plasmiidi sisestamine
bakterisse selekteerimise ja paljundamise
eesmärgil.
Materjalid:
5 µl
Ligeeritud DNA segu
Plasmiid, mis hakkame sisestama
100 µl
E.coli kompetentsed
rakud (TOP10), kompetentsuse aste 107
Rakud, kuhu sisestame plasmiidi
kokku 200ml
Steriilne
tardsööde LB
Kus peal oma rakke kasvatame
200 µl
Ampitsilliini vesilahus 100mg/ml
Selektsioonimarker
25 µl
IPTG 0,8M vesilahus
Värvusreaktsioon/Aktiveerib lacZ promootorit
200 µl
X-gal
Värvusreaktsioon
Arvutused:
X-gal: Tahame
saada 20 µg/ml ja meil on 200ml ehk 20*200=4000 µg ehk 4mg on vaja.
Meil on olemas 20mg/ml lahus, selleks et saada sealt 4mg vütame
1ml/5= 200 µl
IPTG: Tahame
saada 0,1mM=0,0001M ehk 0,0001 = m/0,2 ja sealt saame et vaja minev m
= 0,00002g. Meil on 0,8M vesilahus ehk 0,8 = 0,00002/V ja V = 25 µl
Ampitsilliin:
Tahame saada 100 µg/ml ja meil on 200ml ehk siis 100*200 = 20000 µg
mis on 20 mg. Meil on lahus 100mg/ml ehk siis peame võtma 1000 µl /
5 ehk 200 µl.
Töö käik:
- Meil oli eelnevalt juba ligaas inaktiveeritud ja reaktsioonisegu oli jääl
- Võtame -80kraadi juurest E.coli rakud sulama.
- Transformeerimiseks võtame 100 µl kompetentseid rakke ja lisame reaktsioonisegule.
- Inkubeerime 15 minutit jääl.
- Sulatame 200ml LB söödet, jahutame umbes 50 kraadi juurde (muidu ampitsilliin laguneb kuumuse käes ära) ja lisame sinna X-gal, IPTG ja ampitsilliini. Loksutame segamini ja valame söötme õhukeste kihtidena Petri tassidele.
- Teeme transformatsioonisegule kuumašokki 50sekundit 42 kraadi juures. Meetod seisneb selles, et CaCl2 tõttu DNA tõmbus rakumembraanide ligi. Ja nüüd kui paneme oma segu sooja, siis membraanikahjustuse tagajärjel DNA migreerub raku sisse.
- Tõstame tuubi 5minutiks jääle jahutma
- Järgneb elustamisefaas. Selleks lisame 500 µl vedelat LB söödet ja inkubeerime 30 minutit 37 kraadi juures.
- Tsentrifuugime bakterid põhja
- Eemaldame sööte bakterite pealt
- Suspendeerin, et tekiks ühtlane lahus
- Kannan bakterisegu Petri tassile.
- Kallan klaasterad tassile ja loksutan et bakterid laiali kanduksid.
- Asetame tassid 37 kraadi juurde kasvama .
Mida ja miks
me transformeerime? Millist tulemust ootad ?
Viime enda
vaheplasmiidi bakterisse, et see paljuneks ja et tekiksid
kolooniad .
Vastavalt valged ja sinised. Meie tahame valgeid kolooniaid saada.
Milleks
lisasime antibiootikumi? Mis oleks saanud kui oleksime lisanud
tertatsükliini?
Selleks, et meie
plasmiidis oli ampitsiliini resistentsussait. Kui lisame seda
antibiootikumi, jäävad ellu ainult transformeeritud bakterid. Kui
oleksime lisanud tertatsükliini pole midagi saanud, sest kõik
bakterid oleksid surnud või siis oleks need elus kus on
tertatsükliini resistentsus. Aga need ei ole meie bakterid.
Mis juhtuks,
kui unustaksime lisada X- gali ? Kas viga oleks saanud parandada?
Milleks X-gali on vaja?
Kui me ei oleks
X-gali lisanud, siis ei oleks tekkinud sini-valget
värvuseid . Me ei
oleks saanud kuidagi enda bakterikoloonaid eristada. Viga saaks
parandada hõõrudes X-galiga tassi.
Praktiline töö nr. 7: Minipreparatsioon
aluselise lüüsi meetodil
Eesmärk: Bakterites paljundatud plasmiidi välja puhastamine
Materjalid:1,5 ml
LB
vedelsööde Kasvatame baktereid vedelsöötmes, et neid aereerida. Sisaldab ka antibiootikumi.
Valge bakterikoloonia
200 µl
Lahus E1
Hoiab rakke elus.
200 µl
Lahus E2
Muudab lahuse aluseliseks
200 µl
Lahus E3
Normaliseerib pH
420 µl
Isopropanool Sadestab
200 µl
Etanool Peseme
soolad välja
Töö käik: - Paneme valged kolooniad vedelsöötmesse kasvama. Inkubeerima 37 kraadi juures 16h.
- Tsentrifuugin bakterid põhja
- Eemaldan tuubist supernatandi.
- Re-suspendeerin bakterisademe E1 lahuses.
- Lisan E2 lahuse ja loksutan tuubi
- Inkubeerime 2 minutit, kuni lahus muutub selgemaks.
- Pipeteerime peale E3 lahuse ja raputame segu.
- Tsentrifuugin 5 minutit
- Tõstan supernatandi uude tuubi ja lisan sinne isopropanooli.
- Tsentrifuugin plasmiidse DNA 10 mini.
- Eemaldab supernatandi ja viskan selle ära.
- Pesen sadet etanoolidga.
- Suspendeerin 30 µl MQs.
Mis juhtub
erinevate biomolekulidega aluselises keskkonnas?
Aluselise
keskkonna loob E2 lahus. Selle
tagajärjes bakteriraku membraani
fosfolipiidid moodustavad mitsellid ja
valgud denatureeruvad.
NaOH denatureerib DNA kõrgemat järku
struktuurid .
Kuidas tagada,
et välja puhastuks vaid meid huvitav plasmiidne DNA, mitte bakteri
enda genoomne DNA.Peale E2 lahuse
lisamist ei tohi liiga kaua inkubeerida. Muidu denatureerub ka
plasmiidne DNA mis järgmises
etapis sadeneb.
Laboris puhastati plasmiidset DNA’d, aga saadud plasmiidi kogus tuli liiga
väike. Mida korduskatses teisiti teha?E1 lahuse
lisamise järel tuleb veenduda et lahusesse ei
jääks tükke. Kõik
peab olema piimjas mass. Muidu need tükid ei lüüsu täielikult.
Jälgida, et peale E2 lisamist ei inkubeeriks liiga palju. Jälgida,
et etanooliga pesteks kogemata sadet kaasa ei võta! Kasvatada
baktereid suuremas
mahus .
Praktiline töö nr. 8: Kontroll- restriktsioon
Eesmärk:
Lõigata
puhastatud plasmiidset DNA’d restriktaasidega, et
kontrollida tekkivate DNA fragmentide suuruste järgi, kas puhastatud
plasmiid on õige.
5 µl
Puhastatud plasmiidne DNA
Seda hakkame kontrollima
3,9 µl
MQ
1 µl
Restriktaasi 10x puhver
Luua sobiv keskkond restriktaasi töötamiseks
0,1 µl
EcoRI
restriktaas (lõppkontsentratsioon 0,1U/ µl)
Lõikab DNA’d EcoRI saitidest.
Agaroosgeel
2 µl
6x DNA värv
Arvutused:
Puhver: 10 x 1/10
= 1µl
Restriktaas: 10U/
µl Tahame saada 0,1U/ µl ja meil on 10 µl. 0,1x10 = 1U 1/10 = x/1
ehk x=0,1
Vesi- 10-5-1-0,1
= 3,9 µl
DNA värv: 10 / 5
= 2.
Töö käik: - Pipeteerime kokku DNA, MQ, restriktaari ja puhvri. Inkubeerime 37 kraadi juures 30 minutit.
- Valame agaroosgeeli (meie töö puhul oli see enne juba valmis)
- Peale 30 minutit tsentrifuugin oma kontroll- restriktsiooni, et koguda kokku kaanelt ja seintelst veeaur.
- Lisame 2 µl 6X DNA värvi ja pipeteerime kogu segu agaroosgeeli hambasse.
- Voolutame 150V juures 10-15 minutit. Teeme geelist pildi.
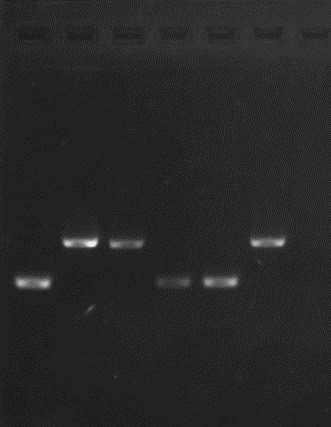
Minu rada: 5
Milleks
teostatakse kontroll-restriktsioon?Et kontrollida,
kas eraldatud plasmiid on õige
Millise
tulemuse sain kontroll-restriktsioonist?
Minu tulemus on
väga ilus, eraldatud plasmiid tuli õige.
Mille järgi valisime sobiva restriktaasi? Veel sobivaid restriktaase.
Valisime EcoRI
restriktaasi, kuna meie insert sisenes pSTBlue-1 vektorisse EcoRV
saiti.
Mõlemalt pool enne seda saiti on EcoRI sait. Seega oli väga
hea valita just see restriktaas, saime mõlemalt poolt ühe
restriktaasiga lõigatud ja vee täpselt väga lähedalt. Võiksime
valida ka muud restriktaasid, nt PstI ja HindIII aga see oleks
tülikas, kuna me ei eraldaks ainult meile huvipakkuvat fragmenti ja
peaksime puhvritega jändama, kuna iga restriktaasi jaoks on erinevat
soolakontsentratsiooni vaja.
Mida silmas
pidada kui kasutada mitut erinevat restriktaasi korraga? Millised
lõikamiseks sobivad tingimused võivad erineda?
Peame vaatama,
mis puhvrit kasutame. Erinevatel restriktaasidel on erinevaid
soolakontsentratsioone vaja. Samuti tuleks jälgida mis temperatuuri
juures need restriktaasid töötavad. Enamusele sobib temperatuus 37
kraadi.
Kontrollrestriktsiooni
geelipildi analüüs.Minu
restriktsioon õnnestus, kuna näeme, et eraldunud
bänd on kuskil
600-700bp, mis peakski nii olema. Rajal ei ole näha teisi segavaid
bände ehk restriktaas ei ole kuskilt mujalt lõiganud.
Praktiline töö nr.9: Bioinformaatiline
sekveneerimise tulemuste analüüs
Eesmärk:
Kloneeritud plasmiidi sekventsi analüüs ja ümberkloneerimise
plaani koostamine
Töö käik ja
analüüs:Mina valisin
DST_T7_E10.ab1
failist sekventsi
Sekventsi
kvaliteedi hinnang: Väga hea kvaliteet, usaldusväärsuse vahemik on
suur ja tausmüra on väga vähe.
Usaldusväärsuse
vahemik : 23-820
Blast ja selle
tulemused:
Identities 632/632 ehk kõik nukleotiidid klapivad ja ei ole toimunud mutatsioone .
Gaps 0/632 ehk ei ole eemaldatud või juurde lisatud kuskile nukleotiide.
Strand Plus/ Miinus ehk sisestatud insert on sisenenud vektorisse tagurpidi ja transkripstsiooni suund on vastupäeva.
Tegemist on
tõepoolest DST geeniga, mis me sisestasime.
Kontrollisin ka
vektori järjestust ja see klapib 100% pSTBlue-1 kloneerimisvektori
järjestusega.
Nukleotiidne
järjestus:
GCGTTACGTATCGGATCCAGAATTCGTGATTTGGATCCTGACATCATTTTGTTAGTGATGGGATGGCCAATCCCTGTGATTACTAATTCACACTGTCGCAGCTGCTGGGCAAACCCCTCATCAACCAGGCCTCTATGCAAAGCTTCGGCCACCCGGTACTTTTTGCCAGTAAGAGGATCAATTATGCCCCCTGTACTGACTTGGGCTTCAAGGCATCGGAGGGCAGTAATCCGATCAACCAAATTTCTACGGGAGGCTTTTAGTACAGAGATCCTTTCCCCACTAGCAGTCAGCCAATACCCTGCAACAGGACTGTGCAAATCTTTCTTGGGGACTAACCGGCTCAGCAAAGAATCAGCAAGTTCTGTAAGTGTGATGAGGCCTTCCTGATACTTTTTGACCAAGGCTTTGTCAATTGTTCCCTGCTCTATAGCCTCATTAATATTGAAGTGTAATCCTGTTTTAGTATCAGTCAGCATATGAGAAGAATGCCCATAGGATTCAAAAAACATAGCTTCCTTCCACTGATATTGCTGCCCTGAAAGTTCAAGATATATACTTTTCTCAATCAGGTTTCTCTGGAAAGCCTCATACACGGTCAATTCTGATCCTGTTTTAGTATCTACTACAGAAATTCTATGTGTTTTCTTGACATTGAGATTGGAAAT
GAA TTC AAA
TCT GAA TTC GTC
GAC
AAGCTTCTCGAGCCTAGGCTAGCTCTAGACCACACGTGTGGGGGCCCGAGCTCGCGGCCGCTGTATTCTATAGTGTCACCTAAATGGCCGCACAATTCACTGGCCGTCGTTTTACAACGTCGTGACT
LEGEND
Kodeeriv ala on boldis ja allajoonitud
Esimene
aminohape
Reverse praimer
Forward praimer
Vektori
järjestus
Milliseid programme kasutasin?
Bioedit näitas
sekveneerimise tulemusi, leidsin sealt usaldusvahemikud ja hindasin
sekventsi kvaliteeti. BLAST andis nukleotiidse järjestuse põhjas
vastava geeni ja seejärel vaatasin mis pidi insert on sisenenud, kas
on gapse ja mutatsioone.
Mis juhtub kui
proovid ühes reaktsioonisegus kahe praimeriga korraga sekveneerida?
Saame kaks
nukleotiidset järjestus mis katavad üksteist ja teevad lugemise
võimatuks. Väga pikkade fragmentide sekveneerimiseks kasutatakse
forward ja reverse praimereid, kuid eraldi reaktsioonides.
Eesmärk:
Sekveneerimistulemuste põhjal koostada plaan funktsionaalse ekspressioonivektori koostamiseks , millega saaks huvipakkuvat valku
(liitvalguna koos GFP-ga) koekultuuri rakuloonides ekspresseerida.
Töö käik:
BamHI
SalI
KASUTAN C1
lugemisraami!
BamHI buffer
BamHI
2- fold excess of
SalI
Incubate at 37°C
Mida on
oluline ümberkloneerimisel silmas pidada?
Inserdis ei tohi
olla ümberkloneerimiseks kasutatave restriktaaside saite, kuna siis
lõigataks meie kodeeritava ala keskelt fragment pooleks, aga me
tahame tervet kodeeritavat ala saada.
Ümberkloneerimist
ei saa teha vaid ühte restriktaasi kasutades, kuna pärast me ei
saaks kontrollida kumba pidi insert vektorisse siseneb. Meil on vaja
aga, et ta õiget pidid siseneks, muidu me ei saa funktsionaalset
valku.
Doonorplasmiid pSTBlue1
vektor koos meie inserdiga
Aktseptor plasmiid pEGDP-C
Reportergeen EGFP
Insert DST
Ümberkloneerimiseks
kasutatavad restriktaasid BamHI ja SalI
Millistel tingimustel restriktsiooni reaktsioon läbi tuleks viia? Millise
pikkusega fragmendid saame ja mida kasutame ligeerimisel?
Double Digest
soovitaks meie reaktsiooniseguks:
- 1x BamHI puhver
- 1x BamHI
- 2x SalI
Incubate at 37°C
Lõigates nii
pSTBlue1 vektorit saame eraldada oma inserdi mis on kuskil 700bp ja
pEGFP-C vektori lõikamisel lõikame sealt kuskil 20 bp välja ja
kasutame ülejäänut pikka vektorit.
Millised
katsed tuleb teostada, et saaksime kätte 1µg sellist GFP ja sinu
kloneeritud valku kodeerivat ekspressioonivektorit?
Lõikame restriktaasidega nii doonor kui akseptor DNA’d. Selleks on vaja BamHI ja SalI restriktaasi ja BamHI puhvrit. Inkubeerime 4h – üleöö 37 kraadi juures
Valame agaroosgeeli ja isoleerime DNA fragmendid agaroosis. Kanname hambasse enda restriktsioonisegu + 6x DNA värvi. Soovime doonorvektorilt saada inserti ja akseptorvektorilt lühikest juppi eemaldada. Selleks lõikame huvipakkuvad fragmendid DNA’st välja ja puhastame DNA agaroosgeelist. Kaalume enda väljalõigatud osa ja lisame sellele ADB puhvrit. Inkubeerime 55 kraadi juures kuni kogu geel on sulanud ja lahus on homogeenne. Seejärel pipeteerime kõik segu ränikolonni, DNA jääb räni külge. Tsentrifuugime. Valame kogumistopsist puhvri ära ja peseme 2 korda pesupuhvriga. Seejärel lisame elueerimispuhvrit ja tsentrufuugime uuesti. Nüüd saime tulemusekt inserdi ja aktseptorplasmiidi.
Järgneb ligatsioon. Võtame vektorit ja inserti suhtes 1:3 ja lisame segule T4 DNA ligaasi ja ligeerimispuhvrit ja MQ’d. Ligeerime 4 kraadi juures üleöö. Kõrvale teeme ka kontroll-proovi mis sisaldab ainult vektor+ligaasi + puhver, ilma inserdita.
Järgmiseks sisestame plasmiidi bakterisse selle paljundamise eesmärgil. Selleks lisan reaktsioonisegule E.coli kompetentseid rakke, inkubeerin 15-30 minutit jääl ja samalajal valan petri tassi LB söötme, kanamütsiili. 30 minuti järel teen kuumašoki 42 kraadi juures 50 sekundit. Lisan LB söödet ja inkubeerin 37 kraadi juures. Kannan bakterid petri tassile ja jätan nad kasvama.
Valin koloonia ja kontrollin kas ligatsioon töötas. Panen koloonia vedelsöötmesse 16ks tunniks kasvama. Seejärel teen miniprepi ehk tsentrifuugin bakterid põhja , eemaldan supernatandi, lisan E1 lahuse, lisan E2 lahuse, Lisan E3 lahuse, tsentrufuugin. Tõstan supernatandi uude tuubi ja lisan isopropanooli. See sadestab ja kallame supernatandi ära. Siis peseme sooladest puhtaks etanooliga. Tsentrifuugime. Kuivatame. Ja lisame vett.
Nüüd meil peaks olema siis õige plasmiid. Kontrollime seda restriktsaasidega lõigates ja pipeteerime segu agaroosgeeli hambasse. Lahutatud fragmendid peaks olema oodatud suurustega 670bp ja 4700 bp.
Tallinna Tehnikaülikool



































Kõik kommentaarid