Termodünaamika seadused ja alused
1.
Kas tegu on avatud, suletud või isoleeritud süsteemiga:
a)
kohv väga hea kvaliteediga termoses; -isoleeritud
b)
jahutusvedelik külmkapi jahustussüsteemis; -suletud
c)
pommkalorimeeter, milles põletatakse
benseeni ; - isoleeritud
d)
automootoris põlev
bensiin ; - suletud
e)
elavhõbe termomeetris; - isoleeritud
f)
taim – avatud
2.
Kirjelda kolme viisi, kuidas saab tõsta siseenergiat avatud
süsteemis! Millisega neist meetoditest saab tõsta siseenergiat
suletud süsteemis? Kas mõni neist meetoditest kõlbab ka
isoleeritud süsteemi energia tõstmiseks?
– avatud
- toimub nii energia- kui ka
ainevahetus ümbritseva keskkonnaga
– suletud
- puudub ainevahetus ümbrusega, aga võib toimuda
energiaülekanne kas töö (mehaaniline toime) või soojusena (
termiline toime).
– isoleeritud
- puudub nii energia- kui ka ainevahetus. Väliskeskkonnaga pole ei
mehhaanilist ega soojuslikku kontakti.
Siseenergiat
avatud süsteemis saab tõsta:
–
paisumistöö :
w = -Pex∆V (töö, mille tulemusena muutub süsteemi ruumala)
– raskuse
tõstmine: w = mg∆h
– elektriline
töö: w = φ∆q
Suletud
süsteemi
siseenergia muutus U üleminekul algolekust lõppolekusse
on võrdne süsteemile antava soojushulga q ja tema heaks tehtava töö
w
summaga . Süsteem võib ka energiat kaotada, st teha tööd või
anda ära mingi osa soojusest. Seega muutub suletud süsteemi
energia.
• Suletud
süsteemi energia muutub tänu energiavahetusele soojuse ja töö
kujul süsteemi ja ümbritseva keskkonna vahel.
Isoleeritud
süsteemi siseenergia ei muutu, kuna puudub soojusülekanne
3.
Protsessifunktsioonid. Energia, töö, soojus . Termodünaamika I
seadus. Olekufunktsioonid . Paisumistöö. Kalorimeetria . Siseenergia.
Nimetage ja seletage termodünaamika esimesest seadusest tulenevaid
järeldusi.
Energia-
keha või jõu võime teha
tööd, džaul
Töö
on liikumine mõjuva jõu
vastu.
Soojus
on energia, mis kantakse üle tänu temperatuuri erinevusele.
Kõrgemalt madalamale.
Termodünaamika
I seadus: isoleeritud süsteemi
siseenergia on
konstantne , mitteisoleeritud süsteemi korral ∆U=q+w
Olekufunktsioonid
on sellised süsteemi olekut iseloomustavad suurused, mis ei sõltu
oleku saavutamise viisist : tihedus, siseenergia.
Konstantse
ruumalaga süsteem ei saa
paisuda,
kui süsteemi ruumala saab muutuda ja väline rõhk on konstantne,
siis w=Pex∆V
Kalorimeetria
– seade, kus soojuse ülekannet mõõdetakse temperatuuri muutuse
kaudu. Koosneb reaktsiooninõust, segajast ja termomeetrist.
Süsteemi
summaarset võimet teha tööd nim tema
siseenergiaks
(U)
4. Entalpia . Soojusülekanne konstantsel rõhul. Soojusmahtuvus . Kumb on suurema molaarse
soojusmahtuvusega,
kas NO või NO2? Miks?
Entalpia
on termodünaamilise süsteemi siseenergia ja rõhuenergia summa.
Soojusmahtuvuseks
nim soojushulka, mis on vajalik antud
ainekoguse temperatuuri
tõstmiseks 1 kraadi võrra.
Soojusmahtuvus
konstantsel ruumalal on
sisenergia muudu ja temperatuuri muudu
jagatis .
Konstantsel
rõhul
aga entalpiamuudu ja temperatuurimuudu jagatis
5.
Kirjutada süsteemi soojusmahtuvuse avaldised püsival rõhul ja
ruumalal. Kummal juhul on
soojusmahtuvus
suurem?
Konstantsel
ruumala:
Konstantsel rõhul:
Soojusmahtuvus
on suurem konstantsel rõhul, kuna osa saadud soojusest kulub
paisumistöö tegemiseks.
6.
Termokeemia, reaktsiooni soojusefekt , endotermiline ja eksotermiline reaktsioon .
Termokeemia-tegeleb
keemiliste muundumiste soojusefektidega, põhineb termodünaamika
esimesel seadusel.
Keemilise
reaktsiooni entalpia on
soojusefekt,
mis kaasneb keemilise reaktsiooniga (kui rõhk ja temperatuur ei
muutu). Entalpiamuut (soojusefekt) sõltub süsteemi alg- ja
lõppolekust, mitte aga protsessi läbiviimise teest või reaktsiooni
vahestaadiumitest!
Eksotermiline
protsess – soojus eraldub
Endotermiline
protsess – soojus neeldub
7.
Miks on paljud eksotermilised reaktsioonid spontaansed? Selgita,
millisel juhul võib endotermiline reaktsioon olla spontaanne!
Paljud
eksotermilised reaktsioonid on spontaansed , kuna
Gibbsi energia
vabaneb ,
entroopia kasvab või kahaneb, endotermiline protsess
võib olla spontaanne juhul, kui entroopia kasvab
8.
Keemilise muundumise entalpia. Reaktsioonientalpia, seos siseenergia
ja entalpia vahel, standardsed reaktsioonientalpiad. Hessi seadus.
Standardsed tekkeentalpiad. Reaktsioonientalpia sõltuvus temperatuurist.
Keemilise
reaktsiooniga kaasneb enamasti ka energia eraldumine või
neeldumine .
Reaktsioonientalpia
on selle vaheetappide entalpiamuutuste summa.
Seos:
konstantne ruumala, konstantne rõhk. Kui reaktsioonis ei osale
gaase , on erinevus ∆U
ja ∆H
vahel väga väike ning ∆U=∆H.
Standardne tekkeentalpia ∆H⁰f
on defineeritud kui 1 mooli aine tekkimisreaktsiooni entalpia,
lähtudes vajalikest elementidest nende kõige stabiilsemates
vormides .
Reaktsioonientalpia
muutust sõltuvalt temperatuuri
muutusest saab arvutada lähtudes ainete soojusmahtuvusest ∆Hr,2º=
∆Hr,1º+
∆CP(T2–
T1)
∆CP
= ΣnCP,m(
produktid )
–
ΣnCP,m(
lähteained )
Hessi
seadus – on füüsikalise
keemia
põhiseadus , mille kohaselt keemilise reaktsiooni soojusefekt
sõltub ainult süsteemi alg- ja lõppolekust, mitte reaktsiooni
käigust
9.
Termodünaamika II ja III seadus.
Termodünaamika
II seadus – ei ole võimalik
selline protsess, kus kogu soojus muudetaks tööks ning pole
võimalik kanda soojust üle külmemalt kehalt soojemale ilma tööd
tegemata.
Termodünaamika
III seadus- Korrapärase
kristallistruktuuriga puhta aine entroopia absoluutsel
nulltemperatuuril on võrdne nulliga. Lühidalt: see seadus selgitab
T = 0 K saavutamise mittevõimalikkust järjestikuste
jahutusprotsesside lõpptulemusena. See seadus annab aluse ainete
absoluutsete entroopiate leidmiseks
10. Carnot ringprotsess . Carnot’ ringprotsessi kui soojusmasina
analüüsist tulenavad järeldused.
Soojusmasina
(Carnot’ ringprotsessi) kasutegur. Soojuspump . Entroopia.
Spontaanne muutus, entroopia ja korrapäratus, entroopiamuut.
Standardsed molaarsed entroopiad ja reaktsioonientroopiad. Globaalne
entroopiamuut. Keskkond. Summaarne entroopiamuut. Entroopia sõltuvus temperatuurist.
Järeldused:
Üks
mool ideaalset gaasi paisub isotermiliselt (A→B) ja adiabaatiliselt
(B→C) ning seejärel surutakse kokku isotermiliselt (C→D) ning
adiabaatiliselt (D→A), nii et gaasi lõppolek vastab algolekule. •
Kõik protsessid on
pöörduvad .
Kasutegur: töö on seda suurem, mida suurem on sooja ja külma keha
temperatuuride erinevus.
Saame
avaldada Carnot’ ringprotsessi
kasuteguri :
Soojuspump: (kui panna Carnot’ protsessid käima
vastassuunas , siis saame
külmutusseadme või soojapumba.
•energia
jäävuse seadus;
•gaasid
kokkusurumisel kuumenevad;
•soojus
kandub alati külmemalt kehalt kuumemale
Entroopia:
Kui
gaas paisub vaakumisse, siis see protsess on iseeneslik.
Vastupidi see protsess ise ei toimu. Carnot’ ringprotsess võimaldab
defineerida uue olekufunktsiooni, mis kirjeldab seda sorti nähtuste
võimalikkust.
(tähistab
qrev
soojuse pöörduvat ülekandmist)
•
Iga protsess, mille jaoks on
∆S>q/T toimub iseeneslikult
•
Iga protsess isoleeritud
süsteemis (∆S>0) toimub iseeneslikult
•
Kui isoleeritud süsteem on
tasakaalus, omab entroopia maksimaalset väärtust.
Spontaanne
muutus: • Iseeneslike protsessidega kaasneb energia ja aine
jaotuse korrapära kahanemine ehk siis korrapäratuse kasv.
•
Entroopia kasvab: sulamisel,
aurustumisel, T-i tõstmisel, gaasi paisumisel, tahke aine
lahustumisel jne
•
Entroopia kahaneb:
veeldumisel, tahkestumisel, gaasiliste ainete mahu vähenemisel:
entroopia
ja korrapäratus: Termodünaamikas mõõdetakse korrapäratust
entroopiaga S. Mida suurem on korrapäratus,
seda suurem on ka entroopia.
Isoleeritud
süsteemis toimuvad iseeneslikud protsessid entroopia kasvu suunas. •
Seega on iga isoleeritud süsteemi
saatuseks muutuda korrapäratuks.
• Pöörduval protsessil suureneb gaasi energia samapalju kui
väheneb soojusallika energia ning süsteemi energia ei muutunud.
– Temperatuuri
tõusuga kaasneb süsteemi korrapära vähenemine, kuna molekulid
hakkavad rohkem (energilisemalt, kiiremini) liikuma.
– Süsteemi
korrapära väheneb ka aine jaotumisel suuremasse ruumalasse või
segunemisel.
Korrapärase
kristallistruktuuriga puhta aine entroopia absoluutsel
nulltemperatuuril on võrdne nulliga.
Entroopiamuut
:Mittepöörduva (spontaanse) protsessi summaarne entroopia muut isoleeritud süsteemis on positiivne. Standardsed molaarsed
entroopiad ja reaktsioonientroopiad.: Standardsed molaarsed
entroopiad
•
Arvestama peab ka, et suurema
temperatuurivahemiku korral sõltub aine soojusmahtuvus
temperatuurist:
S(T)=dT
Standardsed
reaktsioonientroopiad
•
Entroopia tähtsus keemias on
selles, et ta võimaldab ennustada reaktsioonide iseeneslikku
kulgemist või mittekulgemist.
•
Arvestama peab, et
reaktsiooniga kaasnev entroopiamuut koosneb kahest osast:
–
saaduste entroopia erinevus
lähteainetest; – reaktsioonil eralduv soojus tõstab keskkonna
entroopiat .
•
Kui reaktsiooni käigus kulub
või tekib gaas, on vastav entroopiamuut domineeriv ja
reaktsioonientroopia märki saab sellest lähtuvalt ennustada.
Standardne
reaktsioonientroopia ∆Sr on defineeritud kui reaktsioonisaaduste ja
lähteainete molaarsete entroopiate vahe, võttes arvesse
stöhhiomeetriakoefitsiente:
r0=
m0(
saadused )-
m0
(lähteained)
Globaalne
entroopiamuut.: TD 2 seadus, kehtib isoleeritud süsteemi kohta, s.t
sellist süsteemi, kus ei toimu aine ega soojuse vahetust ümbritseva
keskkonnaga, seega tuleb arvesse võtta ka keskkonna entroopia
muutust. Eksotermilise reaktsiooni käigus eraldub keskkonda soojust,
mis põhjustab seal
märgatava entroopia kasvu.
•
Tänu sellele süsteemi
entroopia võib kahaneda ja reaktsioon on spontaanne.
Keskkond:
Isoleeritud süsteemi moodustavad süsteem ise ja teda ümbritsev
keskkond.
Konstantsel
rõhul ja temperatuuril toimuva protsessi korral ('keskkond' on
piisavalt suur) on keskkonna entroopia muutus arvutatav kui: Skeskk
=-
•
Summaarne entroopiamuut
avaldub:
Ssum
= Ssüst
+Skeskk
- Protsess on spontaanne, kui ∆Ssum on positiivne.
•
Seega võib süsteemi
entroopia ka kahaneda, kui selle arvelt keskkonna entroopia vähemalt
sama palju kasvab.
Entroopia
sõltuvus temperatuuris: Reaktsiooni ∆H ja ∆S ei muutu oluliselt
temperatuuri muutudes, kuna temperatuur mõjutab nii lähteainete kui
ka produktide vastavaid suurusi samas suunas.
•
Reaktsiooni
vabaenergia muutub
temperatuuri muutudes aga märgatavalt ja võib isegi märki muuta!
11.
Vabaenergia. Reaktsiooni vabaenergia, vabaenergia ja töö,
temperatuuri mõju vabaenergiale. Millised keemilised reaktsioonid
toimuvad iseeneslikult ja kui palju nad võivad tööd teha?
Vabaenergia-
süsteemi energia, mille arvel ta saab konstantsel temperatuuril ja
rõhul tööd teha
Seega
kulgeb iga protsess iseeneslikult vabaenergia vähenemise suunas (ΔG
Tasakaaluolekus ΔG = 0
Standardne
reaktsiooni vabaenergia:
Reaktsiooni
ΔH ja ΔS ei muutu oluliselt temperatuuri muutudes, kuna temperatuur
mõjutab nii lähteainete kui ka produktide vastavaid suurusi samas
suunas. Reaktsiooni vabaenergia muutub temperatuuri muutudes aga
märgatavalt ja võib isegi märki muuta!
Keemiline
tasakaal
1.
Keemilise reaktsiooni tasakaal-
Kui tingimused ei muutu, kulgevad reaktsioonid olekuni, kus
vastassuunaliste reaktsioonide kiirused saavad võrdseks, ainete
kontsentratsioonid enam ajas ei muutu ja tekkinud segus on sõltuvalt
tingimustest rohkem või vähem kõiki reaktsioonis
osalevaid aineid
= tasakaal
2.
Reaktsiooni pöörduvus, termodünaamika ja keemiline tasakaal,
tasakaalukonstandid,
heterogeenne tasakaal.
3.
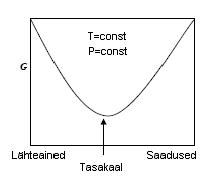
Keemilise tasakaalu tingimused (G abil, teha joonis).
Keemilisele
tasakaalukonstandile vastab Gibbsi vabaenergia miinimum e ∆G=0.
Kui
p ja
T on
const , siis saavad spontaanselt kulgeda vaid need protsessid, mille
käigus Gibbsi energia väheneb (
ΔGP,
T
Kui
ΔG
saab võrdseks nulliga, siis
on süsteem saavutanud tasakaaluoleku ning iseeneslikult sellest enam
väljuda ei saa.
Kui
temperatuur ei ole konstant, siis saab otsustada reaktsiooni suuna
üle reaktsiooni entalpiamuudu ja entroopimuudu koosmõju järgi
vastavalt võrrandileΔ
G =
Δ
H –
TΔ
S4.
Keemilise tasakaalu ja reaktsiooni suuna kriteeriumid (Q ja K abil)
Tasakaalukonstandi väärtus näitab kuidas reaktsioon kulgeb: K suur väärtus (K>1)
=> tasakaalulisesreaktsioonisegus on ülekaalus saadused. K on
väike (K tasakaalulises reaktsioonisegus on ülekaalus
lähteained
5.
Reaktsiooni Gibbsi energia muut. Reaktsiooni
isotermi võrrand (van’t
Hoff ).
6.
Termodünaamiline
tasakaalukonstant ning tema avaldised rõhkude ja
kontsentratsioonide kaudu.
7.
Tasakaalukonstant lahustes ja heterogeensete süsteemide korral.
8.
Kirjeldage Le Chatelier’ printsiibi alusel, kuidas muutub
reaktsioonisegu tasakaaluline koostis, kui lisada või eemaldada
reagente, segu kokku suruda või lasta sellel paisuda või muuta
temperatuuri.
9.
Millised järgmistest väidetest on õiged, millised mitte (kui
valed, siis
põhjenda , miks)?
a)
Tasakaalu saabumisel reaktsioon peatub.
b)
Tasakaalulist reaktsiooni ei mõjuta produktide lisamine.
c)
Kui tõsta reagendi rõhku, siis muutub tasakaalukonstant
suuremaks .
d)
Kui tõsta lähteainete kontsentratsiooni, siis produktide
tasakaalulised kontsentratsioonid
kasvavad.
10.
Millised järgmisetest väidetest on õiged, millised mitte (kui
valed, siis põhjenda, miks)?
a)
Tasakaalulises reaktsioonis hakkab pöördreaktsioon toimuma kohe,
kui on hakanud tekkima
produktid.
VALE, sest reaktsioon peab jõudma enne tasakaaluolekusse (reagendid
tekivad sama kiiresti, kui nad ära tarbitakse)
b)
Kui muuta reaktsioon kiiremaks, siis produktide tasakaalulised
kontsentratsioonid suurenevad. VALE, sest dünaamilist keemilist
tasakaalu iseloomustab see, et mõlemad protsessid kulgevad sama
kiirusega ja reaktsioonisegu üldine koostis ei muutu.
c)
Tasakaaluolekus on reaktsiooni Gibbsi energia võrdne nulliga. ÕIGE
d)
Tasakaaluolekus on reaktsiooni standardne Gibbsi energia võrdne
nulliga. ÕIGE
Kontrollida
üle!
11.
Kc ja Kp konstandid ja avaldised.
Kc
on iseloomulik konstant, mis oleneb temperatuurist, kuid ei olene
reageerivate ainete kontsentratsioonist.
Kp-
osarõhkude kaudu avaldatud ; kui osalevad gaasilised ained.
Mida
suurem on Kc või Kp, seda enam on tasakaalu segus saadusi, st
reaktsiooni tasakaal on nihutatud paremale - saaduste tekke suunas.
12.
Mida näitab tasakaalukonstandi väärtus?
Tasakaaluoleku
matemaatiliseks kirjeldamiseks kasutatakse tasakaalukonstanti K.
Reaktsioonisegu tasakaalulist koostist kirjeldab tasakaalukonstant,
mis võrdub saaduste aktiivsuste korrutise ja lähteainete
aktiivsuste korrutise jagatisega,
kusjuures iga aktiivsus on
astendatud vastava aine stöhhiomeetrilise kordajaga tasakaalustatud
reaktsioonivõrrandis.
13. Koosta järgmiste reaktsioonide tasakaalukonstantide Kc avaldised:
a)
CO(g) + Cl2(g)
⇌
COCl(g) + Cl(g) Kc=[COCl][Cl]/[CO][Cl₂]
b)
2 H2S(g)
+ 3 O2(g)
⇌
2 SO2(g)
+ 2 H2O(g) Kc= [SO₂]2[H2O]2 /[H2S]2[O2]3
14.
Kirjuta reaktsioonijagatis Q:
a)
4 Bi(t) + 3 O2(g)
→
2
Bi2O3 (t) Q= [Bi2O3]2 / [Bi ]4 [O2]3
b)
N2O3 (g)
→
NO(g) + NO2(g)
Q= [NO][NO2]/[N2O3]
Q
– reaktsioonisisalduste kontsentratsioonide (aktiivsuste) korrutis
jagatud reaktsiooni lähteainete kontsentratsioonide korrutisega.
15. Vaatleme reaktsiooni CO(g) + H2O(g)
⇌
CO2(g)
+ H2(g).
a)
Kuidas muutub H2
osarõhk, kui CO2 osarõhku
tõsta? Kui tõsta CO2 osarõhku, siis H2 osarõhk jääb samaks.
b)
Kuidas muutub CO2
osarõhk, kui CO osarõhku vähendada? Kui CO osarõhku vähendada,
siis CO2 osarõhk jääb samaks.
c)
Kuidas muutub H2
kontsentratsioon, kui CO kontsentratsiooni suurendada? Kui CO
kontsentratsiooni suurendada, siis H2 kontsentratsioon suureneb.
d)
Kuidas muutub reaktsiooni tasakaalukonstant, kui H2O
kontsentratsiooni vähendada? Kui H2O kontsentratsiooni vähendada,
siis tasakaalukonstant suureneb.
Rõhu
tõstmisel nihkub reaktsiooni tasakaal suunas, kus gaasiliste ainete
moolide arv on väiksem (seega rõhu vähenemise suunas), rõhu
vähendamisel vastupidi- suurema gaasiliste ainete moolide arvu tekke
suunas (seega rõhu suurenemise suunas). Kui gaasiliste ainete
moolide arvud on aga võrrandi mõlemal poolel võrdsed (või
reaktsioonis ei osale gaasilisi aineid), siis rõhk reaktsiooni
tasakaalu ei mõjuta.
KONTROLLIDA!
16.
Tasakaalulised protsessid (tuua näiteid).
o Tasakaaluline
saab olla ainult niisugune protsess, mis kulgeb lõpmata aeglaselt.
Kuna soojust absoluutselt mittejuhtivaid aineid looduses ei leidu,
siis on
soojushulk , mida süsteem
vahetab ümbruskonnaga, seda
väiksem, mida kiiremini kulgeb protsess. Seega saavad
adiabaatilistele lähedased olla ainult kiiresti kulgevad protsessid.
Niisuguse protsessi näiteks võiks tuua kokkusurumise ja paisumise,
mis toimub gaasi igas punktis
helilaine levimisel.
o Protsess
on tasakaaluline. Kui protsess poleks tasakaaluline, liiguks see
alati vaid stabiilsema seisu poole ning mitte kunagi ebastabiilsema
poole ehk protsess oleks pööramatu
o Dünaamilist
keemilist tasakaalu iseloomustavad järgmised omadused:
1)
ühel ajal toimub nii
pärisuunaline kui ka vastassuunaline protsess;
2)
mõlemad protsessid kulgevad sama kiirusega ja reaktsioonisegu üldine
koostis ei muutu.
Tasakaal
sõltub: rõhust, reagentide lisamisest, temperatuurist.
Reaktsioon,
mis antud tingimustel ei toimu (näiteks hapniku ja vesiniku segu toatemperatuuril ja normaalrõhul), ei toimu ka tingimuste väikese
muutuse korral.
Faaside tasakaal
1.
Heterogeenne süsteem, faas, faasisiire.
Heterogeensed
– koosnevad mitmest erisuguse
omadusega osast (ehk faasist).
Faas
- ühtlase koostisega ja konkreetses olekus olev süsteemi osa, mis
on teistest faasidest eraldatud piirpinnaga. Faas võib koosneda ka
mitmest ainest.
Faasisiire
ehk faasiüleminek- aine üleminek ühest faasist teise (ilma
keemilise koostise muutumiseta).(Nt, jää sulamine,
grafiidi muundumine teemandiks) Faasiüleminekud toimuvad kindla temperatuuri
ja rõhu juures, mis sõltuvad ka aine puhtusest.
2. Aururõhk , aurustumissoojus , sulamissoojus .
Aururõhk
on rõhk, mida avaldab selle kondenseeritud faasiga dünaamilises
tasakaalus olev aur. Aururõhk ongi defineeritud kui vedeliku või
tahkisega tasakaalus oleva auru poolt avaldatav rõhk.
Aurustumissoojuseks
nimetatakse soojushulka, mille peab andma keemistemperatuuril oleva
vedeliku massiühikule, et muuta see sama temperatuuriga auruks.
Aurustumissoojus sõltub temperatuurist ja väheneb temperatuuri
tõustes ning kaob aine kriitilisel temperatuuril. Reeglina
määratakse aurustumissoojus aine keemistemperatuuril normaalrõhul
(nimetatakse ka keemissoojuseks).
Sulamissoojus
on füüsikakonstant, mis näitab aine sulatamiseks kuluvat või
tahkumisel eralduvat energia hulka.
3.
Aururõhu sõltuvus temperatuurist.
Temperatuuri
tõustes vedeliku aururõhk samuti tõuseb, kuni saab võrdseks
atmosfäärirõhuga ja
aurustumine hakkab toimuma kogu lahuse
ulatuses – vedelik hakkab
keema . Kindlal temperatuuril püstitub
vedeliku kohal kindel aururõhk, sõltumata vedeliku hulgast.
Aururõhk sõltub otseselt sellest, kui kerge on molekulil ületada
jõude, mis hoiavad vedelikku koos. Kõrgemal temperatuuril on
molekulidel rohkem energiat ja järelikult peaks ka aururõhk olema
kõrgem.
4.
Aurustumine, kondenseerumine , keemine , külmumine , sulamine ja sublimeerimine .
Aurustumine
– vedeliku üleminek gaasilisse olekusse, energia neeldub.
Kondensatsioon ehk kondenseerumine on aine üleminek gaasilisest agregaatolekust
vedelasse (mõne allika järgi veeldumine) või tahkesse (mõne
allika järgi härmatumine).
Keemine-
kui vedelik aurab
lahtises anumas, siis tekkinud aur levib ruumis ja
tasakaalu ei püstitu. Temperatuuri tõustes vedeliku aururõhk
samuti tõuseb, kuni saab võrdseks atmosfäärirõhuga ja
aurustumine hakkab toimuma kogu lahuse ulatuses – vedelik hakkab
keema. Normaalne
keemistemperatuur on temperatuur, mille juures
vedeliku aururõhk on 1
atmosfäär . Madalamal rõhul toimub keemine
madalamal temperatuuril.
Külmumisel
väheneb vedeliku molekulide energia tasemeni, mil nad enam
liikuda ei saa ja toimub aine üleminek tahkesse faasi. Külmumistemperatuur
on temperatuur, mille juures tahke aine ja vedelik on tasakaalus ning
varieerub sõltuvalt rõhust. Normaalsel külmumistemperatuuril
toimub aine külmumine rõhul 1 atm. Rõhu tõstmisel
külmumistemperatuur reeglina tõuseb. Rõhu tõstmisel vee
külmumistemperatuur langeb.
Sulamine
ja
tahkumine on üleminekud korrapärase struktuuriga tahke oleku
ning korrapäratu struktuuriga, kuid lähedase tihedusega vedela
oleku vahel. Temperatuuri, mille juures tahke ja vedelfaas on
tasakaalus rõhul 1 atm, nimetatakse sulamistemperatuuriks. Antud
aine sulamissoojuseks (sulamisentalpiaks) (ΔHs, kJ/mol) nimetatakse
soojushulka, mis on vajalik 1 mooli aine üleminekuks tahkest olekust
vedelasse konstantsel temperatuuril. Sulamine on endotermiline
protsess, tahkumine eksotermiline protsess. Aine sulatamiseks kulub
energiat (soojust); sulamise käigus soojus neeldub, kuid aine
temperatuur ei muutu.
Sublimeerumine on aine üleminek tahkest olekust gaasilisse ilma vedelat olekut
läbimata. Kergesti sublimeeruvate ainete kohal saab mõõta
aururõhku samuti kui vedelike kohal. Sublimeerumiseks kuluvat
energiat nimetatakse sublimatsioonisoojuseks.
5.
Faaside tasakaal heterogeensetes süsteemides.
(Heterogeenne
süsteem on füüsikalis-keemiline süsteem, mis koosneb vähemalt
kahest faasist.)
Gibbsi
faaside reegel
• Kui
meil on k komponenti (ainet) ja p faasi, siis vabadusastmete
(süsteemi kirjeldamiseks vajalike muutujate) arv f avaldub: f = k –
p + 2
•
Ühekomponentses süsteemis seega f = 3 –
p
•
Ühekomponentses süsteemis ei saa
tasakaalus olla korraga rohkem kui 3 faasi.
Tasakaal
heterogeensetes (2 faasi korraga olemas, aga pole sama koostisega)
süsteemides –
A)ühekomponentne
süsteem – 1 aine, aga vôib mitmes
erinevas agr. olekus olla. Vee
diagramm: vee O e. kolmikpunkt: TO = 0,0078C; pO = 4,6mmHg.
Superkriitiline olek – kaob vahe vedela ja
gaasilise faasi vahel,
tekib tihe ja hästi voolav ollus.
B)
kahekomponentne süsteem – 2 ainet; A ja B. Nii sulamise kui ka
keemise vältes muutub
kummagi faasi koostis eraldi.
6.
Individuaalsete ainete olekudiagrammid ja nende kasutamine.
Faasidiagramm ehk
olekudiagramm on diagramm, kus enamasti temperatuuri ja rõhu
teljestikus kujutatakse süsteemitasakaalulist olekut.
Olekudiagramm
näitab konkreetse aine eelistatud agregaatolekut antud tingimustel.
Olekudiagramm
on tavaline arvutiteaduse mõiste, mida kasutatakse lõplike
automaatide graafilisel kujutamisel. Olenevalt konkreetsest
kasutusalast on seda kirjanduses
käsitletud eri
viisidel .
7. Kriitilised omadused: keemistemperatuur, sulamistemperatuur ,
kolmikpunkt, kriitiline temperatuur ja kriitiline rõhk.
Keemistemperatuur
on temperatuur, mille juures vedeliku aururõhk on 1 atmosfäär.
Madalamal rõhul toimub keemine madalamal temperatuuril.
Temperatuuri,
mille juures tahke ja vedel faas on tasakaalus rõhul 1 atm,
nimetatakse sulamistemperatuuriks.
Kolmikpunktis
saavad kokku 3 faasipiiri ja seal on need 3 faasi omavahel ka
tasakaalus. Kolmikpunkt on ainele iseloomulik suurus, mille
asukohta ei saa muuta.
Kriitiline
temperatuur – temperatuur, mille korral auru ja vedeliku tihedus on
võrdsed (ei saa eristada vedelat ja gaasilist faasi).
Aururõhk
kriitilises temperatuuril – kriitiline rõhk
Kriitilisel
temperatuuril ja rõhul kaob
piirpind auru ja vedeliku vahel ning
gaas (aur) pole enam rõhu tõstmisega veeldatav. Auru veeldamiseks
piisab rõhu tõstmisest, gaasi veeldamiseks tuleb aga ka
temperatuuri alandada.
8.
Superkriitiline olek.
Aine,
mille rõhk ja temperatuur on tema kriitilise punkti väärtusest
kõrgemad
Keemiline kineetika
1.
Reaktsiooni kiirus. Sõltuvus kontsentratsioonist, kiiruste seadused,
reaktsiooni järk.
Reaktsiooni
kiirus on defineeritud kui
lähteaine kadumise või saaduste tekke kiirus.
Enamike
reaktsioonide kiirus väheneb nende kulgemise käigus vastavalt
sellele, kuidas lähteainete
kontsentratsioonid
vähenevad. Seega keskmise kiiruse valemis peaksid kasutatud
kontsentratsioonid olema mõõdetud suhteliselt
lühikese ajavahemiku
tagant, et saadud kiirus iseloomustaks protsessi võimalikult
täpselt.
Reaktsiooni
järk on suurus, mis on
arvuliselt võrdne kontsentratsioonide astmenäitajate summaga
reaktsiooni kiiruse võrrandis.
2.
Mis on keemilise reaktsiooni järk? Milline on reaktsiooni kiirus
0.-ndat ja 1.-st järku reaktsioonides (graafikute abil)? Mis on
iseloomulik 2. järku reaktsioonidele (võrdlus 1. järku
reaktsiooniga)?
Nulljärku
reaktsiooni korral avaldub reaktsiooni kiirus kui v=k, st, kiirus ei
sõltu reagentide kontsentratsioonidest.
Esimest
järku reaktsiooni korral on
reaktsiooni kiirus proportsionaalne lähteaine
kontsentratsiooniga .
Teist
järku reaktsiooni korral on
reaktsiooni kiirus proportsionaalne lähteainete kontsentratsiooni
ruudu või kahe lähteaine kontsentratsioonide korrutisega.
Reaktsiooni
järk on suurus, mis on arvuliselt võrdne kontsentratsioonide
astmenäitajate summaga reaktsiooni kiiruse võrrandis.
3.
Keemilise reaktsiooni kiirus, reaktsiooni kiirust mõjutavad tegurid.
Keemilise
reaktsiooni kiirus näitab, kuidas reaktsiooni komponentide
kontsentratsioon või rõhk ajas muutub.
Reaktsiooni
kiirus sõltub paljudest faktoritest.
Temperatuur
mõjutab sageli keemilise reaktsiooni kiirust üsna suuresti, kuna
kõrgemal temperatuuril on molekulidel suurem
soojusenergia . Kuigi
kõrgemal temperatuuril on põrked molekulide vahel sagedasemad, on
sellest tingitud reaktsioonikiiruse kasv siiski tühine. Kaugelt
olulisem on tõik, et temperatuuri tõusuga kasvab molekulide hulk,
millel on piisav energia reageerimiseks ehk mille energia ületab
aktivatsioonienergiat
Keemilise
reaktsiooni kiirus sõltub reageerivatest ainetest.
Hape -alus
reaktsioonid, soolade moodustumine ning
ioonvahetus on kiired
reaktsioonid. Reaktsioonid, kus molekulide vahel tekib
kovalentne side ja moodustuvad suured molekulid, on reeglina väga aeglased.
Reageerivate molekulide sidemete tugevus ja iseloom mõjutavad
tugevasti produktide tekke kiirust. Vähemate sidemete
ümberpaigutumistega reaktsioonid kulgevad kiiremini kui
reaktsioonid, mille käigus tekib või katkeb rohkem keemilisi
sidemeid .
Reageerivate
ainete agregaatolek mõjutab samuti suuresti reaktsioonikiirust. Kui
reagendid on samas faasis, nagu vesilahusepuhul, viib
soojusliikumine molekulid kokku. Kui aga reagendid on erinevates
faasides , on
reaktsioon piiratud vaid kokkupuutepinnaga ja tugev
segamine võib
osutuda vajalikuks reaktsiooni lõpule
viimiseks . See tähendab, et
mida kõrgema peensusastmega on
reageeriv tahkis või vedelik, seda
suurem on tema eripind ja seda ulatuslikum on tema
kokkupuude teise
reagendiga ning seda kiirem on seega ka reaktsioon
Kontsentratsioon
mängib keemilistes reaktsioonides olulist rolli, sest vastavalt
aktiivsete põrgete
teooriale peavad kaks molekuli reaktsiooni
toimumiseks põrkuma.
Emma -kumma reagendi kontsentratsiooni tõusuga
kasvab ka põrgete sagedus, kuna reageerivaid molekule on
ruumalaühikus rohkem ja nad on pidevalt teineteisele lähemal, mis
toob kaasa reaktsioonikiiruse kasvu.
Lahuse
segamine kiirendab samuti keemilist reaktsiooni,
andes lahuse
osakestele suurema kineetilise energia, mis toob kaasa põrgete arvu
kasvu reagentide vahel, mis omakorda suurendab aktiivsete põrgete
tõenäosust.
Katalüsaator on aine, mis kiirendab keemilist reaktsiooni, kuid mida ei kulu
reaktsiooni käigus. Katalüsaator kiirendab reaktsiooni, võimaldades
reaktsioonil toimuda teistsuguse mehhanismiga, mille
aktivatsioonienergia on madalam. Autokatalüüsi puhul on reaktsiooni
produkt ise toimuvale reaktsioonile katalüsaatoriks, põhjustades
positiivset tagasisidet. Proteiine, mis
toimivad biokeemilistes
reaktsioonides katalüsaatoritena, nimetatakse ensüümideks.
Katalüsaator ei mõjuta keemilise reaktsiooni tasakaalu, ta
kiirendab päri- ja vastassuunalisi reaktsioone võrdselt
Gaasilise
reaktsiooni korral suurendab rõhu tõstmine põrgete arvu
reageerivate ainete vahel, mille tulemusel reaktsioon
kiireneb . See
on
seletatav tõsiasjaga, et gaasilise aine aktiivsus on
proportsionaalne tema osarõhuga.
Tasakaal.
Keemiline kineetika tegeleb keemiliste reaktsioonide kiirustega,
samas kui termodünaamika määrab ära reaktsiooni toimumise
ulatuse . Pöörduva reaktsiooni korral püstitub keemiline tasakaal
hetkel, kui päri- ja vastassuunaliste reaktsioonide kiirused saavad
võrdseks ning reagentide ja produktide kontsentratsioonid enam ei
muutu.
4.
Reaktsiooni kiiruse sõltuvus temperatuurist, van’t Hoffi reegel ja
Arrheniuse empiirilised võrrandid .
Pea
kõik reaktsioonid kulgevad kiiremini kõrgemal temperatuuril.
Arrheniuse võrrand
Van’t
Hoffi seadus ütleb, et mitteelektrolüüdi lahjendatud lahustes on
osmootne rõhk võrdeline lahustnud aine molaarse kontsentratsiooniga
ja temperatuuriga. ,
5.
Katalüsaatori mõju. Homogeense ja heterogeense katalüüsi näiteid.
Katalüsaator
on aine, mis kiirendab reaktsiooni, ise reaktsiooni käigus ära
kulumata.
Heterogeense
katalüüsi korral on
katalüsaator erinevas faasis võrreldes reagentidega. Nt õli ja
vesi
Homogeense
katalüüsi
korral on katalüsaator reagentidega samas faasis, tavaliselt
lahustatuna samas solvendis.
2SO2 +
2NO2 = 2SO3 + 2NO
2NO
+ O2= 2NO2
2SO2
+ O2 + (NOx ) =2SO3
Lahused1.
Lahused ja segud , küllastunud lahus.
Lahus
on kahest või enamast
komponendist koosnev
homogeenne süsteem.
Küllastunud
lahus on lahus, milles on
lahustunud maksimaalne võimalik kogus lahustunud ainet.
Segu
on kahe või enama aine
koosesinemine, kus komponendid pole omavahel keemiliselt ühinenud.
2.
Lahuse kontsentratsiooni väljendamisviisid.
Molaarne
kontsentratsioon- näitab lahustunud aine moolide arvu ühes
kuupdetsimeetris (ühes liitris) lahuses.
Molaalne
kontsentratsioon- näitab lahustunud aine moolide arvu 1
kilogrammis
lahustis .
Moolimurd-
näitab lahustunud aine moolide arvu suhet lahusti ja kõikide
lahustunud ainete moolide arvu summasse. Kui lahus koosneb
lahustist ja vaid ühest lahustunud ainest, siis
- Massimurd, Massi- või mahuprotsent jne.
3. Lahustuvus . Lahustumise molekulaarne olemus, rõhk ja gaaside
lahustuvus ( Henry seadus), temperatuur ja lahustuvus,
lahustumisentalpia ja entroopia.
Lahustuvus–
küllastatud lahuse kontsentratsioon.
Molaarne
lahustuvus on aine molaarne
kontsentratsioon küllastunud lahuses.
Henry
seadus : gaaside lahustuvus
konstantsel temperatuuril on proportsionaalne nende osarõhkudega:
C=KHP.
Kõrgemal
temperatuuril toimub ainete
lahustumine reegline kiiremini. Gaaside
lahustuvus kõrgemal temperatuuril on madalam. Enamus tahkete ainete
lahustuvus kasvab temperatuuril tõustes.
Lahustumisentalpia
on 1 mooli aine lahustumisega kaasnev entalpia muutus.
Entroopia
kasvab korrastatud tahkise lahustumisega.
4.
Ideaalsete lahuste aururõhk: Raoult `I seadus.
- Raoult’i seadus: lahusti aururõhk on proportsionaalne tema
moolimurruga lahuses: Plahusti=
PpuhasXlahusti
5.
Lahuste kolligatiivsed omadused: aururõhu alanemine,
keemistemperatuuri tõus ja külmumistemperatuuri alanemine, osmoos
ja pöördosmoos.
Kolligatiivsed
omadused – rida lahuste
omadusi, mis sõltuvad ainult lahuse kontsentratsioonist, mitte aga
lahustunud ainest.
Aururõhu
alanemine: lahuse
moodustumisega kaasneb entroopia kasv ja tulemusena lahuse
vabaenergia kahanemine. Seega peab lahusega tasakaalus oleva auru
vabaenergia samuti vähenema ja seetõttu ongi aururõhk väiksem.
Lahuse
aururõhu suhteline langus on võrdne lahustunud aine moolimurruga
lahuses:
,
kus .
Lahustumisega
kaasneb entroopia kasv ja selle tulemusena ka lahuse vabaenergia
kahanemine, seega peab ka auru vabaenergia kahanema ning sellele
vastab
keemistemperatuuri tõus.
Külmumistemperatuur alaneb , kui lisada lahusesse
nt soola. Lahustunud ained alandavad vedeliku külmumistemperatuuri.
Et lahuse aururõhk on madalam kui lahustil, saabub
külmumistemperatuurile vastav vedela ja tahke faasi aururõhkude
võrdsus lahuse korral madalamal temperatuuril. Lahusti ja lahuse
külmumistemperatuuride vahet nimetatakse külmumistemperatuuri
languseks: .
Osmoos
on nähtus, kus
solvent tungib läbi poolläbilaskva membraani
kontsentreeritumasse lahusesse, kusjuures lahusti liigub madalama
kontsentratsiooniga lahusest (vee puhul kõrgem veepotentsiaal)
lahusesse, kus on kõrgem lahustunud aine kontsentratsioon (vee puhul
madalam veepotentsiaal).
Pöördosmoos
on nähtus, kus lahust liigub läbi poolläbilaskva membraani
lahustunud aine väiksema kontsentratsiooni suunas.
6.
Jaotusseadus. Destillatsioon, ekstraktsioon ja kromatograafia.
Jaotusseadus
: lahustunud aine
kontsentratsioonide suhe kahes tasakaalus olevas lahuses on püsiv
suurus.
Kromatograafia
on
üldmõiste mitmesuguste
laboratoorsete füüsikalis-keemiliste meetodite kohta, mida
kasutatakse uuritavate ainete segu komponentide lahutamiseks paljude
sorptsiooni ja desorptsiooni tingimustes.
Ekstraktsioon
on meetod lahustunud ainete
segude lahutamiseks. Lahust segatakse
temas mittelahustuva teise solvendiga. Lahustunud ained jaotuvad kahe
vedela faasi vahel.
Destillatsiooniks
nimetatakse vedeliku aurustamist
keetmisel ja sellele
järgnevat kondenseerimist vastuvõtjasse.
Kolloidkeemia ja pinnanähtused
1.
Pihussüsteemide (dispergeeritud) mõiste ja klassifikatsioon ,
faasidevaheline
piirpind.
Pihussüsteem
e dispersne süsteem on füüsikalises keemias kahe-või
enamafaasiline süsteem, kus
pihustunud aine asub
dispersioonikeskkonnas e üks aine on jaotunud teises. Jaotuvad :
jämedispersne süsteem, kolloiddispersne süsteem ja tõelised
lahused.
- Aerosoolid: vedelik või tahke aine on jaotunud gaasilises keskkonnas (udu, suits); ei saa olla kõrge kontsentratsiooniga
- Vahud: jämepihused (vedel keskkond õhukeste kiledena gaasimullide vahel) (vahukoor); ei ole agregatiivselt püsivad ja lagunevad teatud aja jooksul
- Emulsioon: vedelik on jaotunud vedelikus ; erinevad polaarsused (piim, koor, kreemid, lakid )
- Suspensioon : jämepihused (savi, tsement , värvid, pastad)
- Soolid ehk kolloidlahused : kolloidosakese suurusjärgus tahked osakesed jaotunud vedelikus
Tarded ja geelid:
makromolulaarsete ühendite lahused ja kolloidsüsteemid
2.
Kolloidlahused.
Kolloidlahused
on sellised heterogeensed lahused, mis silmaga vaadates
tunduvad ühtlased. Seega segades liiva vette me kolloidlahust ei saa, sest
liivaterad on palja silmaga nähtavad. Osakeste suurus kolloidlahuses
on 1...100 nm. Kolloidlahused ei ole termodünaamiliselt stabiilsed.
See tähendab seda, et aja jooksul kolloidlahus laguneb näiteks
sademe tekke näol.
3.
Kolloidsüsteemid ja kolloidosakesed toiduainetes (piim ja piimatooted ).
Kolloidsüsteemid
on laialt levinud nii looduses kui ka tehnikas. Organismides
kulgevate eluprotsesside ja tekkivate struktuuride aluseks on
valgud ,
tärklis, tselluloos jt. suuremolekulilised ained. Toiduained (leib,
või, margariin, koor, liha), riided ja jalatsid (
kiudained , nahk,
kumm , plastmassid jt.) moodustavad mitmesugust tüüpi
kolloidsüsteeme.
4.
Emulsioonid, nende stabiliseerimine , emulgaatorid
Emulsioonid
on süsteemid, mis sisaldavad kahte vedelfaasi, millest üks on
peenete tilkadena dispergeeritud teises. Termodünaamiliselt
ebapüsivad. Kahe segunematu vedelingrediendi, näiteks vee ja
vedela parafiini, kokku
segamisel ja intensiivsel loksutamisel on
võimalik saada ebapüsiv emulsioon. Kui üks faas jaotub teises
väikeste gloobulitena, siis tulemuseks on piirpinna suurenemine ja
pinna vaba energia suurenemine süsteemis. Saadud seisund on
termodünaamiliselt ebastabiilne ning selle tagajärjel diperssest
faasist tekivad sfäärilised
tilgad (sfäärilisel
osakesel on
minimaalne piirpind sama ruumala korral) ja need tilgakesed liituvad,
põhjustades faasi eraldumise ning süsteem saavutab minimaalse vaba
energia. Pindaktiivse aine lisamisel adsorbeeritakse see piirpinnale
ja õ/v piirpinna aktiivsus väheneb, emulgeerimise protsess
paraneb ja stabiilsus süsteemis samuti.
5.
Tyndalli efekt.
Kuna
kolloidlahuses on pihustunud aine osakesed tunduvalt suuremad kui
tõelises lahuses, siis on need osakesed nähtavad pihust läbivas
valguses. Nii tekib valguse läbijuhtimisel kolloidlahuses silmaga
nähtav valguskiirte tee, tõelises lahuses aga mitte.
6.
Kolloidosakese ehitus, kolloidosakese laengu tekitamine.
Kolloidosakene
on küllalt
keeruka ehitusega, koosnedes paljudest ioonidest
(molekulidest, aatomitest). Osakese ehituses võime eraldada kahte
osa, seesmist - neutraalset ja selle ümber välist - ionogeenset
osa. Sisemine osa - tuum koosneb aine molekulidest või aatomitest
ning moodustab kolloidosakese põhilise massi.
Laengu
teke: Tuum adsorbeerib oma pinnale kas positiivseid või negatiivseid
ioone, mis annavad tuumale laengu. Tuuma pinnale adsorbeeruvaid ioone
nimetatakse potentsiaali määravateks ioonideks. Tekkinud laengu
tõttu tõmbuvad tuuma vahetusse lähedusse
vastasmärgilised ehk nn.
vastasioonid. Potentsiaali määravad
ioonid koos vastasioonidega
moodustavad kolloidosakese välise ehk ionogeense osa (vt.
disperssete süsteemide
elektrilised omadused).
7.
Kolloidsüsteemide jaotus, lüofiilsed ja lüofoobsed süsteemid,
mitselli moodustumine pindaktiivsete ainete lahustes.
Jaotus:
Jämedisperssed-, kolloiddisperssed- ja molekulaardisperssed
süsteemid.
Lüofoobsed süsteemid. Selliste süsteemide korral on dispersioonikeskkonna ja dispersse faasi osakeste vahelised molekulaarjõud suhteliselt nõrgad. Vesikeskkonna korral nimetatakse neid süsteeme ka hüdrofoobseteks süsteemideks.
Lüofiilsed süsteemid. Nende süsteemide korral on dispersioonikeskkonna ja dispersse faasi osakeste vahelised mõjujõud küllaltki suured. Vesikeskkonna korral nimetatakse neid süsteeme hüdrofiilseteks süsteemideks.
Vastas
ioonid on tuumaga seotud erinevalt. Osa vastasioone on seotud
vahetult tuuma pinnaga ning nad moodustavad koos potentsiaali
määravate ioonidega elektrilise kaksikkihi adsorbse osa.
Ülejäänud
vastasioonid, mis on tuumaga nõrgemini seotud seetõttu, et neid
mõjustab keskkonna soojuslik liikumine, moodustavad elektrilise
kaksikkihi difuusse osa.
Tuum
koos adsorbse kihiga moodustab graanuli ning graanul koos difuusse
kihiga mitselli. Dispersioonikeskkonda koos selles sisalduva
elektrolüüdiga nimetatakse intermitsellaarseks vedelikuks.
8. Pindaktiivsed ained.
pindaktiivsete
ainete molekulid koosnevad polaarsest rühmast ja mittepolaarsest süsivesinik ahelast, siis püüavad nende ainete molekulid omandada
adsorbses kihis teatud kindla orientatsiooni. Pindaktiivne
aine on keemiline
aine,
millel on võime vähendada vee
ja teiste vedelike
või tahkiste
pindpinevust ,
suurendades ühtlasi nende märgumist.
Levinuimad pindaktiivsed
ained on seebid ,
detergendid,
dispergendid,
sünteetilised pesemisvahendid .
9.
Kolloidsüsteemide stabiilsus.
Termodünaamiliselt
ebastabiilne dG = s ds, e. dG on negatiivne kui pindala
väheneb.
Seega peab
püsivus seisnema kineetilistel iseärasustel. Võib ka öelda et
kolloidsüsteemid on termodünaamiliselt labiilsed, aga kineetiliselt
stabiilsed. Tõmbuvad atraktsioonijõudude toimel, mis vähenevad
ligikaudu võrdeliselt 1/R2 kui R on osakeste vaheline
kaugus. Nendel interaktsioonidel palju pikem ulatus kui
üksikaatomite korral. Vastu töötavad näiteks kaitsev kile,
samamärgilised laengud osakeste pinnal jne
10.
Agregatiivne püsivus, koagulatsioon , koagulatsioonilävi ja selle
arvutamine, Shultze- Hardy reegel.
Agregatiivne
püsivus väljendub osakeste otseses võimes säilitada dispergeerimisastet. Süsteem on alati
valmis vähendama oma eripinda (s.t. agregeeruma). Seega peab kogu
aeg olema seda takistav
ületamatu potentsiaal. Selleks on
agregatiivse püsivuse korral pinnalaeng (üks ja sama, siis
tõukuvad) ja
solvatatsioon (takistab kokkupuudet, võib ka olla
laengute tõukumine). Peab ületama solvatatsioonienergia.
Koagulatsioon
on kolloidosakeste agregeerumine suuremateks osadeks . Sellele
tavaliselt järgneb väljasadestumine.Koagulatsioonil on kaks
staadiumi: 1)Varjatud staadiumis disp. aste väheneb, kuid
suurenenud osakesed pole nähtavad. 2) Nähtavas staadiumis on
muutused silmaga nähtavad (suured tükid, hägune või sademe
tekkimine)
Koagulatsiooni
kutsub esile see ioon , mille laeng on vastasmärgiline osakese
laengule ning suurema laenguga iooni koaguleeriv toime on suurem
(Schulze-Hardy
reegel).!
Koagulatsioonilävi
on selline elektrolüüdi kogus, mille juures hakkab märgatav koaguleerimine . Sellega on seotud kriitiline tseeta-potentsiaal,
millest allpool hakkab koaguleerumine.
11.
Faasidevaheline piirpind, protsessid piirpinnal .
Suuri
osakesi moodustavad ka kõrgmolekulaarsed ühendid (edaspidi
tähistame lühidalt - kmü), millede molekulid - makromolekulid -
koosnevad samuti sadadest ja tuhandetest aatomitest ning on
mõõtmetelt samuti küllaltki suured. Niisugustes süsteemides on
juba üsna märgatav faasidevaheline piirpind. Seetõttu tuleb
sellistes süsteemides pöörata tähelepanu ka faasidevahelisel
piirpinnal kulgevatele nähtustele.
12.
Pinna vabaenergia. – Pinna
vabaenergia on töö, mis tuleb teha pinna suurendamiseks ühe
pindalaühiku võrra. dw = s ds,
kus dw – tehtud töö, s – pindpinevus ( J*m-2 e.
N*m-1, kuna J = 1N*m ), ds – pindala muutus
13.
Adsorptsiooni mōiste
Adsorptsioon on pinnanähtus, mille puhul vedeliku või gaasi molekulid kogunevad
molekulaarjõudude toimel tahke keha pinnale.
14.
Gibbsi adsorptsioonivōrrand.
T=
c/RT * δσ/δc kus, c – PAA kontsentratsioon lahuses, σ –
pindpinevus vedelik-gaas pinnal, Γ - adsorbeeritud aine liig pinnakihis
15.
Adsorptsioon vedeliku ja gaasi piirpinnal. Adsorptsioon on pinnanähtus, mille puhul vedeliku või gaasi
molekulid kogunevad molekulaarsidejõudude (van der Waalsi jõudude) toimel tahke keha pinnale.
16.
Mis on adsorptsioon? Kuidas seda liigitatakse?
•Füüsikalise
adsorptsiooni aluseks on füüsikalised nähtused – van der Waalsi
jõud adsorbaadi osakeste vahel.
•Kemosorptsioonil
tekib keemiline side adsorbendi ja adsorbaadi vahel. Võib tekkida
nii elektroni üleminekul kui ühise elektronpaari tekkel.
(Liigitatakse
: hapniku sisaldavad ühendid, süsiniku põhised ühendid, polümeeride põhised)
17. Absorptsioon ja adsorptsioon (erinevus)
Adsorptsioon
on pinnanähtus, mille puhul vedeliku või gaasi molekulid kogunevad
molekulaarsidejõudude (van der Waalsi jõudude) toimel tahke keha
pinnale. Absorptsioon on gaasi või gaasisegu neeldumine vedelikus
või tahkises (vedeliku neeldumist tahkises).
18.
Millised ained on hüdrofoobsed , millised hüdrofiilsed ?- hüdrofoobne - vett tõrjuv. Hüdrofiilne – vett omastav.
Hüdrofoobsed ained on nt paljud metallid ja teatud orgaanilised
ained (molekulid ei sisalda polaarseid aatomirühmi). Hüdrofoobsed
ained lahustuvad apolaarsetes/orgaanilistes lahustites . Hüdrofiilsus
ehk veelembus on aine võime vastastikuliseks mõjuks veega.
Hüdrofiilsed ained on näiteks anorgaanilised soolad , tärklis ja savid .
19.
Langmuiri võrrand. Langmuiri
adsorptsiooniisotermi on esitatav kujul Γ = Γmc/(k+c)
Kus,
Γ - adsorptsioon (mol/cm2), Γm – adsorptsiooni suurus pinna
täielikul küllastumisel adsorbeeruva ainega
(maksimaalne
adsorptsioon), c – aine kontsentratsioon lahuses, k -
tasakaalukonstant.
20.
Adsorptsioon tahke aine ja lahuse piirpinnal.
Adsorptsioon
on pinnanähtus, mille puhul vedeliku või gaasi molekulid kogunevad
molekulaarsidejõudude toimel tahke keha pinnale.
21.
Mis on pindpinevus?-
Pindpinevus ehk pinna vabaenergia töö, mis tuleb teha pinna
suurendamiseks ühe pindala ühiku võrra. Nähtus, kus pinnakiht
käitub kui elastne kile.
22.
Vedeliku pindpinevuse määramise meetodid.
1. Stalagnomeetriline:
Loetakse kindlast ruumalast tekkinud tilkade arvu.
2. Rõnga
lahtirebimise meetod: Pindpinevuse saab määrata otseselt jõust,
mida läheb vaja rõnga lahtitõmbamiseks vedeliku pinnalt.
3. Rehbinderi
meetod (suurima rõhu meetod): Siin mõõdetaks rõhku, mida on vaja
rakendada, et suruda läbi kapillaari ava ühe vedeliku sisse teise
vedeliku tilk või gaasimullike.
23.
Mis on pindliig ?
Gibbsi
adsorptsiooniks e. Gibbsi pindliiaks nimetatakse aine hulka, mis
tuleb lisada süsteemile, kui pindala suureneb ühe ühiku võrra
(näiteks 1 cm2) selleks, et aine kontsentratsioon süsteemis jääks samaks.
24.
Kuidas määrata pindliiga pindpinevuse isotermi alusel?
Süsteemi
komponendi pindliiga Γ ehk absorbeerunud aine hulka defineeritakse
pindkihi ja faasi sisemuse kontsentratsioonide vahena pinnaühiku
kohta.
Saab
määrata Gibbsi adsorptsioonivōrrandiga:
Γ=
- c/RT x ∂σ/∂c kus, c – PAA kontsentratsioon lahuses, σ –
pindpinevus vedelik-gaas pinnal, Γ - adsorbeeritud aine liig
pinnakihis. Järeldused sellest võrrandist:
1. Kui
pindpinevus suureneb kontsentratsiooni kasvades dσ/dc>0, siis Γ
































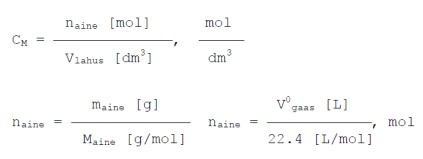
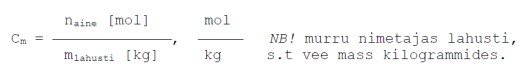

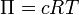
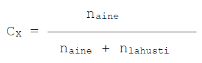

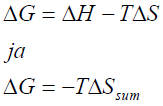
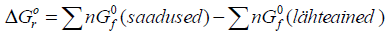
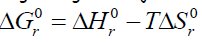
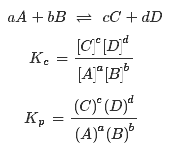
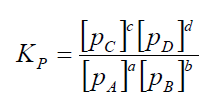
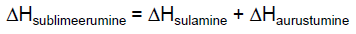




Kõik kommentaarid