VESI
Vee struktuur ja omadused
Tänu meie igapäevasele kogemusele
tunduvad vee omadused meile
tavalised , keemiliste
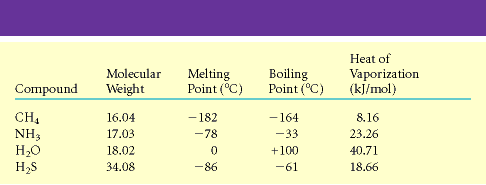
ühendite hulgas on vesi aga üks ebatavalisemaid. Tabel 3.1 toob võrdlevalt välja vee ja mõnede
sarnase molekulmassiga ühendite füüsikalised omadused. Enamikul sarnastel
madalmolekulaarsetel ühenditel on madal keemispunkt ja nad on normaalrõhul ja
toatemperatuuril gaasilised ained. Mis teeb vee nii eriskummaliseks? Vastus peitub
veemolekulide
omaduses moodustada omavahel vesiniksidemeid.
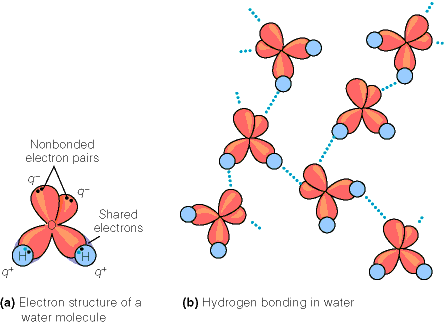
Veemolekuli elektronstruktuur on skemaatiliselt toodud joonisel 3.1 a. Hapnikuaatomi
kuuest välise elektronkihi orbitaalidel paiknevast elektronist kaks on kaasatud kovalentsete
sidemete moodustamisse kahe vesinikuaatomiga. Ülejäänud neli elektroni esinevad kahe vaba
elektronpaarina ja need elektronpaarid on suurepärased vesiniksideme aktseptorid. Samas
käituvad veemolekuli koostises olevad –OH rühmad kui vesiniksideme doonorid. Seega on iga
veemolekul ühtlasi nii vesiniksideme aktseptoriks kui ka doonoriks ja vesi koosnebki omavahel
vesiniksidemetega ühendatud veemolekulide võrgustikust (joonis 3.1b). Sellest tulenevalt
(
vesiniksidemete lõhkumiseks kulub energia) ongi veel oma molekuli suurust arvestades
erakordselt kõrge
keemistemperatuur ja suur
aurustumissoojus (tabel 3.1). Tulenevalt kõrgest
keemistemperatuurist on vesi enamikus Maa pinnal valitsevast temperatuurivahemikust vedelas
olekus.
Vee üleminekuga tahkesse faasi (jäätumine) kaasneb veemolekulide vaheliste
vesiniksidemete korrapära kasv. Jäätumise tulemusena moodustub jäik tetraeedriline
molekulaarne võre, kus iga veemolekul on vesiniksidemete kaudu ühendatud nelja naaber
veemolekuliga (joonis 3.2a). Jää sulamisel laguneb molekulaarne võre ainult osaliselt ja teatud
korrapära säilib ka isegi vedelas olekus ja suhteliselt kõrgetel temperatuuridel. Vee struktuuri
võib vaadelda pidevalt vahelduvate vesiniksidemete kaudu ühendatud veemolekulide
kogumitena, „vahelduvate klastritena”, kus on säilinud osaliselt jää kristallvõre jäänukid, mis on
molekulide soojusliikumise tõttu pidevas katkemise ja reformeerumise protsessis (joonis 3.2c).
Jää kristallvõre küllaltki „avatud” struktuurist tuleneb veel üks vee omapära. Jää sulamisel saavad
veemolekulid asetseda teineteisele lähemal ja seetõttu on vee tihedus (erinevalt enamikust
keemilistest ühenditest) vedelas olekus suurem kui tahkes olekus. See vee omadus on kriitiline
elu eksisteerimise
seisukohast Maal. Kui vesi oleks oma omadustelt sarnane enamikule ainetele ja
omaks tahkes faasis suuremat
tihedust kui vedelas, siis vajuks veekogu pinnale tekkiv jää põhja.
Seal, isoleeritult ülemiste kihtide poolt, hakkaks jää aegade jooksul akumuleeruma põhjustades ka
suurte veekogude täielikku läbikülmumist.
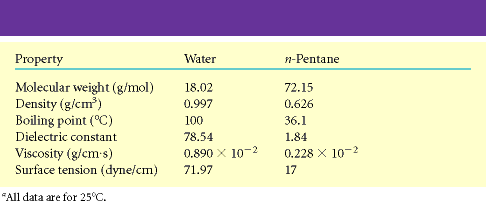
Ka teised vee eripärad (tabel 3.2) on seletatavad lähtudes vee molekulaarsest struktuurist.
Erinevalt enamikust orgaanilistest vedelikest on vesi kõrge viskoossusega. Vee kõrge
viskoossus tuleneb vees esinevatest
eelpool mainitud vahelduvatest vesiniksidemete vahendatud
korrapärastest struktuuridest. Nii
imelikult kui see ka ühe vedeliku kohta ei kõla, tuleb öelda, et
vesi on struktuuriga vedelik. Kohesiivsus (molekulaarjõudude poolt põhjustatud seos ühe ja sama
aine molekulide vahel) on
vastutav ka vee suure pindpinevuse eest. Lisaks on veemolekuli
polaarsusest tingitud ka vee kõrge dielektriline konstant,
D =
ca 80 (näitab, mitu korda on
elektrostaatiline interaktsioon vees nõrgem kui
vaakumis ). Kõrge dielektriline konstant ütleb
meile, et veemolekuli dipoolid on võimelised efektiivselt varjestama
vesikeskkonnas paiknevaid
laenguid.
1
Vesi kui solvent
Eluprotsesside
toimimine nõuab, et osavõtvad molekulid saaks vabalt ringi
liikuda .
Selleks peavad nad olema lahustunud kujul. Vesi on universaalne solvent (lahusti) nii raku sise-
kui väliskeskkonnas. Vee kui solvendi omaduste eest vastutavad kaks juba eelpoolmainitud
veemolekulide omadust: võime moodustada vesiniksidemeid ja
polaarne iseloom. Aineid, mis
interakteeruvad veemolekulidega ja lahustuvad vees hästi, nimetatakse
hüdrofiilseteks (vett
armastav)
aineteks .
Hüdrofiilsed molekulid vesilahuses
Molekulid, mis sisaldavad vesiniksideme moodustumist võimaldavaid funktsionaalseid
rühmi, kalduvad moodustama vesiniksidemeid vee molekulidega. Funktsionaalrühmad, mis
soosivad vees lahustumist on näiteks karboksüül, karbonüül, hüdroksüül, amino, karbamoüül,
sulfhüdrüül jne. Kui ained, mis sisaldavad intramolekulaarseid (molekulisiseseid)
vesiniksidemeid vees lahustada, siis võivad osad (või kõik) intramolekulaarsed
vesiniksidemed asenduda vesiniksidemetega veemolekulide osalusel.
Kuid vees ei lahustu mitte ainult vesiniksidemete doonorid ja aktseptorid. Erinevalt
enamikust orgaanilistest solventidest on vesi heaks lahustiks ioonsetele ühenditele. Näiteks
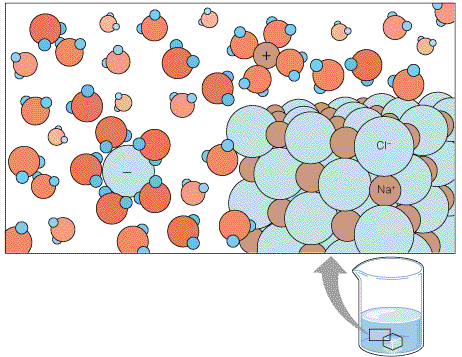
söögisool NaCl, mis esineb tahkel kujul stabiilse ioonvõrega kristallina, lahustub vees hästi.
Vastus peitub veemolekulide polaarsuses. Veemolekulide dipoolid interakteeruvad katioonide ja
anioonidega põhjustades viimaste
hüdraatumist. Hüdraatunud
ioon on ümbritsetud veemolekulide
kihtide poolt (
hüdratatsiooni kihid ) (joonis 3.3). Paljude
ioonsete ühendite nagu NaCl vees
lahustuvus on tagatud kahe faktori poolt. Esiteks on hüdratatsioonikihtide moodustumine
energeetiliselt soodne. Teiseks, nähtuvalt vee kõrgest dielektrilisest konstandist varjestab vesi
efektiivselt laenguid ja seega on ioonilist kristalli koos hoidvad elektrostaatilised
interaktsioonid vesikeskkonnas oluliselt nõrgestatud. Veemolekulide polaarse iseloomu tõttu lahustuvad vees ka
sellised mitteioonsed kuid polaarsed orgaanilised ühendid nagu amiidid,
fenoolid ja estrid.
Hüdrofoobsed molekulid vesilahuses
Hüdrofiilsete molekulide vees lahustuvus põhines nende energeetiliselt soodsal
interaktsioonil veemolekulidega. Seega pole üllatav, et ühendid nagu süsivesinikud, mis ei ole
võimelised moodustama vesiniksidemeid ja ei ole ka polaarsed, lahustuvad vees ainult väga
piiratud koguses. Selliseid molekule nimetatakse
hüdrofoobseteks (vett kartvad). Erinevalt
hüdrofiilsetest molekulidest ei moodustu hüdrofoobsete molekulide ümber hüdratatsiooni kihti.
Vette asetatud hüdrofoobse molekuli ümber tekib hoopis veemolekulidest
regulaarne , jää-sarnane
klatraatstruktuur, mis moodustab hüdrofoobse molekuli ümber nii öelda „puuri”. Hüdrofoobset
molekuli ümbritsev klatraatstruktuur võib olla küllaltki ulatuslik ja põhjustab lokaalset
organiseerituse kasvu vee struktuuris. Igasugusele korrapära kasvule vastab süsteemi madalam
entroopia nii, et hüdrofoobse molekuli asetamine vette põhjustab vee entroopia languse (Δ
S on
negatiivne ja -
TΔ
S on positiivne ning see teeb Δ
G positiivseks). Vee entroopia langus on üheks
hüdrofoobsete molekulide lahustumatuse põhjuseks, isevooluliselt tahaksid veemolekulid olla
võimalikult organiseerimata ehk juhuslikult. Vee entroopia langus on ka üheks põhjuseks, miks
hüdrofoobsed molekulid omavad vesilahuses
tendentsi agregeeruda. Õli vette asetamisel ja
loksutamisel tekivad vette õlitilgad. Ühe suure tilga ümber moodustunud veemolekulide klatraadi
tekkega kaasneb väiksem korrapära kasv kui on kahe väiksema tilga ümber moodustunud
klatraatide põhjustatud
summaarne korrapära kasv. Teisisõnu öeldes, vette asetatud hüdrofoobne
aine püüab minimiseerida oma pindala ehk minimiseerida kontakti veega. Kõikvõimalikest
geomeetrilistest kujunditest omab antud ruumala juures kõige väiksemat pindala kera ja seetõttu
püüavadki vette asetatud hüdrofoobsed ained võtta kera kuju.
2
Amfipaatsed molekulid vesilahuses
Ühe olulise ja huvitava ühendite klassi moodustavad ühendid, millel on
samaaegselt nii
hüdrofiilsed kui
hüdrofoobsed omadused. Selliseid ühendeid nimetatakse
amfipaatseteks.
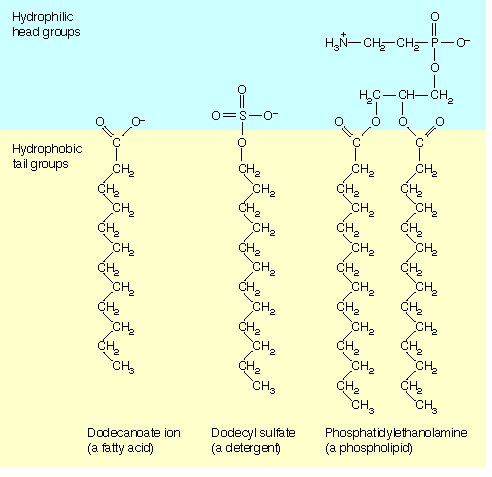
Tüüpilisteks amfipaatseteks ühenditeks on
rasvhapped ja
detergendid (joonis 3.4). Amfipaatse
rasvhappemolekuli hüdrofiilset osa nimetatakse
peaks ja hüdrofoobset osa
sabaks. Sabaks on tihti
pikk süsivesinikahel. Amfipaatsete ühendite segunemisel veega võivad neist moodustuda
erinevad
struktuurid . Esiteks võivad nad vee pinnale moodustada monomolekulaarse n.ö.
üksikkihi, kus pea osad on
kontaktis veega ja sabad ulatuvad veest välja. Kui amfipaatse ühendi
ja vee segu hoolega
loksutada , siis võivad moodustuda kerajad struktuurid nagu
mitsellid ja
kahekihilised
vesiikulid. Mitsellid on molekulide üksikkihist koosnevad kerajad struktuurid, mille
sisemusse jäävad amfipaatsete molekulide sabad,
kusjuures pead asetsevad kera välispinnal ja on
kontaktis veega. Kahekihiliste vesiikulite puhul on kerajas struktuur moodustunud amfipaatsete
molekulide kaksikkihist ja osa veemolekule on kera sees „lõksus”. Soodustamaks omavahelisi
van der Waalsi interaktsioone püüavad hüdrofoobsed süsivesinik sabad alati hoiduda teineteisele
nii lähestikku kui võimalik. Polaarsed või ioniseeritud pead aga on kontaktis veega, kus nad on
hüdrateeritud kujul.
Biokeemia seisukohast kõige olulisem on amfipaatsete molekulide osalemine
bioloogiliste membraanstruktuuride moodustamisel. Bioloogiliste membraanide kaksikkihid, mis
ümbritsevad rakke ja rakusiseseid organelle, koosnevad valdavalt amfipaatsetest molekulidest.
Ioontasakaalud
Väljaarvatud mõningad membraanide hüdrofoobses sisekeskkonnas aset leidvad
reaktsioonid, toimuvad kõik
biokeemilised reaktsioonid vesikeskkonnas. Rakus ja ka rakuvälises
vesikeskkonnas esinevad
ioonid nagu K+, Cl- ja Mg2+, aga ka paljud molekulid ja makromolekulid
mis sisaldavad ioniseeritavaid gruppe. Nende ühendite käitumine biokeemilistes reaktsioonides ja
protsessides on tihtipeale otseselt määratud nende kooseisus olevate ioniseeritavate gruppide
ioniseerituse
vormiga . Järgnevalt vaatamegi ioontasakaalu mõningaid
aspekte keskendudes
eeskätt happe-aluse
vahelisele tasakaalule. Kogu järgnev jutt käib vesilahuste kohta.
Happed ja alused: prootoni doonorid ja aktseptorid
Brønstedi hapete ja aluste teooria järgi on
happed ühendid, millel on
kalduvus loovutada prootonit ja
alused on ained, millel on
kalduvus liita prootonit. Keemias on kasutuses veel üks
üldisem happe-aluse teooria (Lewise teooria) kuid kuna biokeemias on enamik happeid ja aluseid
just Brønstedi happed ja alused, siis jääme me selle teooria raamesse. Tugev
hape dissotsieerub
peaaegu täielikult prootoniteks ja vastavateks anioonideks. Näiteks on HCl peaaegu täielikult
dissotsieerunud H+ ja Cl- ioonideks ja seeläbi tekkinud H+ ioonide hulk vastab HCl hulgale.
Sarnaselt on
NaOH tugev alus, kuna ta ioniseerub täielikult Na+ ja OH- ioonideks,
viimased on
aga väga tugevad prootoni aktseptorid.
Siiski on enamik biokeemias ette tulevatest hapetest ja alustest nõrgad happed ja nõrgad
alused, mis dissotsieeruvad ainult osaliselt. Nõrga happe vesilahuses esineb alati tasakaal happe ja
vastava konjugeeritud aluse vahel.
Konjugeeritud alus on ühend, mis tekib vastavast
happest prootoni loovutamise tagajärjel. Kui konjugeeritud alus liidab prootoni, siis tekib tagasi algne
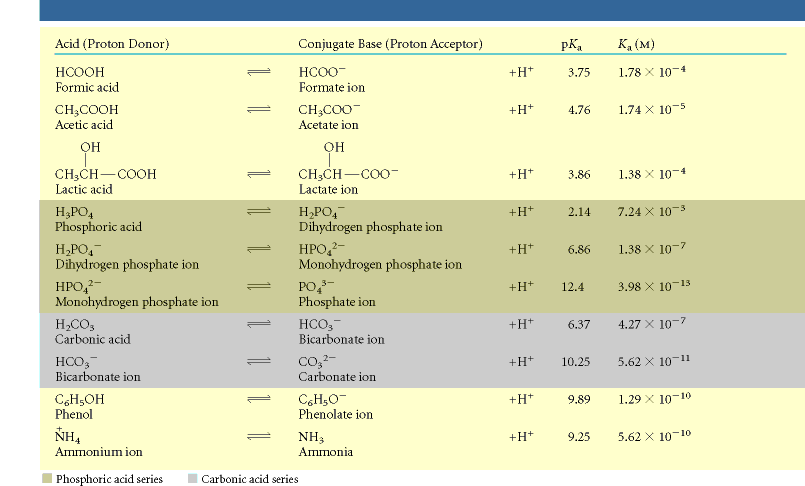
hape. Mõningad nõrkade hapete esindajad koos vastavate konjugeeritud alustega on toodud
tabelis 3.3.
Tabelis 3.3 toodud happed varieeruvad suuresti oma happe tugevuse poolest, s.t. oma
tendentsilt loovutada prootonit. Happe tugevust iseloomustab tema
dissotsiatsioonikonstant Ka ja
selle
negatiivne kümnendlogaritm pKa. Mida tugevam on hape, seda nõrgem alus on vastav
3
(temast tekkinud) konjugeeritud alus ehk mida kergemini loovutab hape prootoni, seda viletsam
prootoni
aktseptor on ka vastav konjugeeritud alus.
Vee ionisatsioon, vee ioonkorrutis
Kuigi vesi on oma põhiolemuselt
neutraalne ühend omab ta siiski kerget tendentsi
loovutada prootonit. Tegelikult võib vesi käituda samaaegselt nii väga nõrga happe kui väga
nõrga
alusena (mitte segi ajada konjugeeritud alusega). Kõige paremini tuleb see ilmsiks kui me
vaatame ionisatsioonireaktsiooni toimuvana nii, et üks veemolekul annab prootoni üle teisele
veemolekulile. Tulemuseks on
hüdrooniumiooni H3O+ ja
hüdroksüüliooni OH- moodustumine.
Seega võib vesi käituda nii prootoni
doonori kui aktseptorina:
H2O + H2O ↔ H3O+ + OH- (3.1)
See on siiski lihtsustatud vaatenurk kuna ülekantav
prooton võib olla assotsieerunud erinevate
veemolekulide klastritega moodustades ioone üldvalemiga H
5O2 , H7O3 jne. Vesilahuses on
prooton äärmiselt mobiilne ja positiivne laeng hüppab ühelt veemolekulilt teisele keskmiselt
ligikaudu iga 10-15 sekundi järel. Kuigi prooton ei esine vesilahuses kunagi H+ kujul
piisab praktilisteks rakendusteks ka ionisatsiooni vaatlemisest lihtsustatud kujul. Enamasti on täiesti
piisav kui kirjutame vee ionisatsiooni üles järgmiselt:
H2O ↔ H+ + OH- (3.2)
Siin on
soovitav siiski meeles pidada, et vesilahuses ei ole prooton kunagi eraldiseisev vaid kui
me räägime prootonist vesilahuses, siis me mõtleme hüdreeritud prootonit. Eeltoodud tasakaal on
kirjeldatav
tasakaalukonstandi K abil:
K = [H+][OH-]/[H2O] (3.3)
Vee kontsentratsioon on alati ligikaudu
konstantne suurus, ligikaudu 55,6 M. Meenutame, et
molaarne kontsentratsioon on aine moolide arv ühes liitris lahuses (mol/l ehk M). Kuna vee
molaarmass on 18 g/mol ja tihedus ca 1000 g/l, siis on ühes liitris ehk 1000 grammis 1000/18 =
55,6 mooli vee molekule, mis võrdubki vee molaarse
kontsentratsiooniga . Võttes võrrandis 3.3
konstantsed liikmed kokku saame uue konstandi,
Kw =
K [H2O], mida nimetatakse
vee ioonkorrutiseks. Asendades
Kw võrrandisse 3.3 saame:
Kw = [H+][OH-] = 1 x 10-14 M2 (t = 25ºC)
(3.4) Kuna vee ioonkorrutis
Kw on konstantne suurus (konstantsel temperatuuril), siis ei saa vesilahuses
[H+] ja [OH-] varieeruda teineteisest sõltumatult. Kui me muudame kas [H+] või [OH-] lisades
vette kas hapet või alust, siis peavad [H+] ja [OH-]
muutuma kooskõlastatult. Kui üks kasvab, siis
teine peab vähenema vastavalt nii, et nende korrutis oleks alati jääv (
Kw). Kõrge vesinikioonide
kontsentratsiooniga [H+] lahuses on madal hüdroksüülioonide kontsentratsioon [OH-] ja
vastupidi.
Kui me vaatame puhast vett, kuhu pole lisatud ei happelisi ega aluselisi ühendeid, siis
peavad kõik H+ ja OH- ioonid pärinema vee dissotsiatsioonist, järelikult:
[H+] = [OH-] = √10-14 M2 = 1 x 10-7 M (kui t = 25ºC)
(3.5) 4
Lahuse kohta, kus [H+] = [OH-] öeldakse, et lahus on
neutraalne, s.t. et lahus pole ei happeline
ega aluseline. Kuna
Kw sõltub temperatuurist, siis ei ole neutraalses lahuses alati [H+] = [OH-] =
1,0 x 10-7 M. Näiteks 37ºC juures on neutraalses lahuses [H+] = [OH-] = 1,6 x 10-7 M.
pH, füsioloogiline pH vahemik
Vältimaks ebamugavat negatiivsete 10 astendajatega opereerimist avaldatakse vesinikioonide
kontsentratsioon pH kaudu. pH on defineeritud järgnevalt:
pH = -log [H+] (3.6) Rangemalt võttes on pH defineeritud kui negatiivne kümnendlogaritm vesinikioonide
aktiivsustest, kuid lahjades lahustes kehtib väga hästi ka seos 3.6 . Mida kõrgem on [H+], seda
madalam on vastava lahuse pH, madal pH vastab happelisele lahusele. Kui lahuses on [H+]
madal, siis peab seal, vastavalt seosele 3.4, olema kõrge [OH-]. Kõrgele pH väärtusele vastab
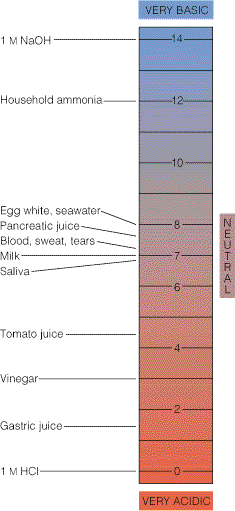
aluseline lahus. pH väärtuste skaala koos mõningate tavalisemate lahuste pH väärtustega on
toodud joonisel 3.5. Enamiku kehavedelike pH väärtused jäävad vahemikku
6,5 – 8,0 ja seda
nimetatakse
füsioloogiliseks pH vahemikuks . Valdav enamus biokeemilisi protsesse toimub just
selles pH vahemikus. Kuna paljud biokeemilised protsessid on äärmiselt tundlikud isegi väikeste
pH kõikumiste suhtes, siis on pH kontrollimine biokeemilises praktikas väga oluline.
Nõrkade hapete ja aluste tasakaal
Paljud bioloogiliselt olulised ühendid sisaldavad oma koostises ioniseeritavaid gruppe,
milleks on valdavalt kas nõrgad happed või nõrgad alused. Suured valgumolekulid sisaldavad
arvukalt happelisi (näit karboksüülgrupp) või aluselisi (näit aminogrupp) ioniseeritavaid gruppe.
Nende gruppide ioniseerituse vorm sõltub keskkonna pH väärtusest ja seega on ka valgu kui
terviku funktsioneerimine pH sõltuv. Heaks näiteks on siin ensüümid, mis omavad tihtipeale
aktiivsust ainult teatud gruppide õige ionisatsiooni vormi puhul ja seetõttu on paljude ensüümide
aktiivsus pH sõltuv. Järgnevalt
vaatleme lähemalt nõrkade hapete ja aluste dissotsiatsiooni
tasakaalu.
Ka ja pKa Kõik tabelis 3.3 toodud reaktsioonid on vaadeldavad happe dissotsiatsioonina, mille
tulemusena eraldub prooton ja tekib konjugeeritud alus. Dissotsiatsiooni tasakaalu üldisel
vaatlemisel kirjutame eraldi välja ainult eemalduva prootoni ja märgime juurde ühendi laengu,
ülejäänud molekuli osa ehk konjugeeritud aluse tähistame tähega A. Sõltuvalt lähteühendi
laengust võib dissotsiatsioonireaktsiooni üles märkida järgmiselt:
HA+ ↔ A + H+
HA ↔ A- + H+
HA- ↔ A2- + H+
Lähtuvalt laengu jäävuse seadusest peab laengute summa olema reaktsioonivõrrandi mõlemal
pool võrdne.
Pange tähele, et konjugeeritud alus võib olla negatiivse laenguga või laenguta, aga
igal juhul peab tal olema üks positiivne laeng vähem kui vastaval happel.
Mugavuse mõttes
kirjutame edaspidi kõik sellised reaktsioonid üles kujul:
5
HA ↔ A- + H+ (3.7)
Nõrga happe dissotsiatsiooni tasakaalukonstant Ka, mida nimetatakse reeglina lihtsalt
dissotsiatsioonikonstandiks, on defineeritud kui:
Ka = [H+][A-]/[AH] (3.8) Mida suurem on happe
Ka väärtus, seda kergemini ta dissotsieerub ja seda suurem on antud happe
tendents loovutada prootonit ning seda tugevam on hape. Kuna nõrkade hapete
dissotsiatsioon on
suhteliselt väheulatuslik, siis on ka vastavad
Ka väärtused väikesed (tabel 3.3). Analoogselt
vesinikioonide kontsentratsiooniga on ka siin tavaks esitada dissotsiatsioonikonstant tema
negatiivse kümnendlogaritmi, pKa kaudu:
pKa = -log Ka (3.9) Kuna
pKa on
Ka negatiivne kümnendlogaritm, siis vastab väikesele
Ka väärtusele suurem
pKa väärtus ja vastupidi. Seega, mida tugevam on hape, seda väiksem on tema
pKa väärtus ja mida
nõrgem on hape, seda suurem on tema
pKa väärtus. Mõningate hapete
Ka ja
pKa väärtused on
toodud tabelis 3.3. Mõned happed nagu
fosforhape ja süsihape võivad loovutada rohkem kui ühe
prootoni (tabel 3.3). Selliseid happeid nimetatakse
polüprootseteks hapeteks. Iga prootoni
loovutamine on vaadeldav eraldiseisva
etapina ja igale etapile vastab temale omane
pKa väärtus
(tabel 3.3). Seega võivad polüprootsed happed esineda lahuses erinevate omavahel tasakaalus
olevate ioonsete vormidena.
Faktorid , mis mõjutavad hapete dissotsiatsiooni
Konkreetse happe dissotsieerumise tendentsi määrab ära dissotsiatsiooni soodustavate ja
pärsivate faktorite omavaheline tasakaal. Mõningad hapete dissotsiatsiooni (
pKa –d) mõjutavatest
faktoritest on seletatavad lähtuvalt vee kui solvendi omadustest. Happe dissotsiatsioonile järgneb
prootoni ja (juhul kui konjugeeritud alus on negatiivse laenguga) negatiivse laenguga
konjugeeritud aluse hüdratatsioon veemolekulide poolt. Kuna hüdratatsioon on energeetiliselt
soodne ja lisaks aitab varjestada laenguid, siis võib öelda, et enamikul juhtudel soodustab
hüdratatsioon hapete dissotsiatsiooni. Erandi moodustavad siin positiivse laenguga happed nagu
NH +
4 , mille dissotsiatsioonil tekib laenguta konjugeeritud alus. Siin stabiliseerib hüdratatsioon
happelist (protoneeritud) vormi ja see on ka põhjuseks, miks NH +
4 on nii nõrk hape.
Dissotsiatsiooni pärsiva faktorina võib nimetada soodsat elektrostaatilist
interaktsiooni negatiivselt laetud konjugeeritud aluse ja positiivse laenguga prootoni vahel. Dissotsiatsioon
nõuab erimärgiliste laengute teineteisest eraldamist. Elektrostaatilise interaktsiooni efekti võime
näha kui võrdleme tabelis 3.3 toodud fosforhappe erinevate ioonsete vormide dissotsiatsiooni
pKa väärtusi. Konjugeeritud aluse negatiivse laengu suurenedes väheneb vastava happe
pKa väärtus.
Kui fosforhape loovutab esimese prootoni
pKa väärtusega 2,14 siis kolmas prooton eraldub alles
pK2-
a väärtusega 12,4 (HPO4 on juba väga nõrk hape). Keskkonnaefektide ja läheduses
paiknevate laetud gruppide mõju hapete dissotsiatsioonile tuleb hästi ilmsiks ka valkudes
paiknevate sarnaste ioniseeritavate gruppide
pKa väärtuste küllaltki suures varieeruvuses.
Nõrkade hapete tiitrimine : Henderson -Hasselbalchi võrrand
Kuidas muutub lahuse pH, kui me muudame seal nõrga happe [konjugeeritud alus]/[hape]
suhet? Küsimuse võib esitada ka teistpidi, kuidas sõltub nõrga happe [konjugeeritud alus]/[hape]
suhe lahuse pH-st? Need on biokeemikule olulised küsimused, kuna paljud rakus esinevad
väikesed molekulid ja makromolekulid sisaldavad nõrgalt ioniseeruvaid gruppe. Mõlemale
6
küsimusele saame vastuse kui võtame nõrga happe dissotsiatsioonikonstandi
avaldise (3.8)
mõlemast poolest negatiivse kümnendlogaritmi. Pärast mõningat reorganiseerimist saame:
-log[H+] = -log
Ka + log([A-]/[AH]) (3.10)
Asendades -log[H+] = pH ja -log
Ka =
pKa saame
Henderson-Hasselbalchi võrrandi, mis seob
omavahel lahuse pH ja [konjugeeritud alus]/[hape] suhte:
pH = pKa + log([A-]/[AH]) (3.11) Näiteks sipelghappe puhul näeb võrrand 3.11 välja järgmiselt:
pH = 3,75 + log([HCOO-]/[
HCOOH ]) (3.12)
Teades lahuse pH-d (näiteks mõõtes pH
meetriga ) saame leida [konjugeeritud alus]/[hape] suhte
(antud temperatuuril on
pKa konstantne suurus). Kui meil on aga teada eeltoodud suhe, siis võime
lihtsalt arvutada vastava lahuse pH.
Henderson-Hasselbalchi võrrandi rakendus ilmneb selgelt tiitrimistulemuste
kirjeldamisel. Selle põhjal saab täpselt arvutada, kuidas muutub lahuse pH kui happe lahusele
lisada teatud kogus alust või vastupidi. Oletame, et me tahame tiitrida 1M sipelghappe lahust
naatrium hüdroksiidiga. Kõigepealt peame küsima, milline on lahuse pH kui meil on 1 mool
sipelghapet 1 liitris lahuses? Seda on võimalik leida vastavalt seosele (3.8). Siin peame eeldama,
et valdav enamus prootoneid tuleb sipelghappe, mitte vee dissotsiatsioonist (arvestades
sipelghappe 1M kontsentratsiooni on see igati õigustatud). Kuna ühe sipelghappe molekuli
dissotsiatsioonil
tekkib üks prooton H+ ja üks HCOO-, siis võime kahe viimase kontsentratsioonid
tähistada ühtmoodi,
z. Kui [H+] = [HCOO-] =
z siis:
Ka = 1,78 x 10-4 = ([H+][HCOO-]/[HCOOH])=
z2/(1-
z) (3.13)
Täpse vastuse saamiseks peaksime lahendama ruutvõrrandi. Nõrkade hapete puhul on aga suurus
z reeglina palju väiksem happe üldkontsentratsioonist (meie näites 1M) ja seega võib murru
nimetajas oleva suuruse võtta ligikaudu võrdseks happe üldkontsentratsiooniga. Meie näite puhul
kus happe üldkontsentratsioon oli 1M võime kirjutada:
Ka ≈ z2 ja
z = [H+] = [HCOO-] = 1,33 x 10-2 M (3.14)
Pange tähele, et ainult ligikaudu 1% sipelghappest on dissotsieerunud ja seega oli meie
lihtsustatud lähenemine õigustatud. Kui meil oleks olnud tegemist lahjema sipelghappe lahusega,
siis see nii ei pruugiks olla, lahjendamine soodustab hapete dissotsiatsiooni. Väga lahjade
happelahuste puhul tuleb juba arvestada ka vee enda ionisatsiooni (arvake, milline on 10-10 M HCl
lahuse pH?).
Eelnev arvutus ütleb meile, et 1M sipelghappe lahuse pH on ligikaudu 1,9. Mis juhtub
kui me hakkame nüüd sellele lahusele lisama NaOH lahust? Lisatud NaOH dissotsieerub
täielikult Na+ ja OH- ioonideks. Kuna vesilahuses on hüdroksüülioonid prootonitega tasakaalus
vastavalt seosele
Kw = [H+][OH-], siis peab OH- lisamisel prootonite hulk lahuses vähenema.
Prootonite eemaldamine kutsub esile sipelghappe edasise dissotsiatsiooni seadmaks jalule
võrrandi 3.13 poolt määratud suhet. Selle tulemusena kaasneb NaOH lisamisega paralleelselt ka
suhte [HCOO-]/[HCOOH] pidev suurenemine. Tulenevalt Henderson-Hasselbalchi võrrandist
(3.11) peab sellega omakorda kaasnema ka lahuse pH pidev tõus. Tiitrimise keskpunktis on pool
algsest sipelghappest NaOH poolt neutraliseeritud. See tähendab, et pool sipelghappest on oma
7
algses happelises vormis ja pool on konjugeeritud alusena ja järelikult [HCOO-] = [HCOOH] ehk
([A-]/[HA]) = 1. Henderson-Hasselbalchi võrrand lihtsustub nüüd
kujule :
pH =
pKa + log 1 =
pKa (3.15)
Seega on nõrga happe tiirimiskõvera keskpunktile vastava lahuse pH võrdne happe
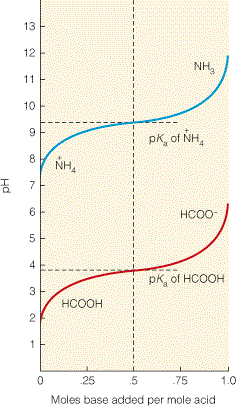
pKa väärtusega. Eeltoodu on leidnud hulgaliselt eksperimentaalset kinnitust. Joonisel 3.6 on toodud
kahe nõrga happe, sipelghappe ja ammooniumiooni tiitrimiskõverad. Joonisel 3.6 on y-
teljel lahuse pH ja x-teljel lisatud aluse moolide arv ühe algselt lahuses olnud happemooli kohta. Pange
tähele, et tiitrimiskõvera
laias vahemikus jääb lahuse pH ühe ühiku piiresse, kas allapoole või
ülespoole
pKa väärtust, pH väärtusel pH =
pKa on tiitrimiskõver kõige laugem. Tiitrimine on
täielikult pöörduv (ja kiire). Meie näites NaOH-ga lõpuni tiitritud sipelghappelahus on täpselt
sama joont pidi tugeva
happega (näiteks HCl) tagasi tiitritav.
Puhverlahused
Vaadates joonisel 3.6 toodud tiitrimiskõveraid
selgub , et kui lahuse pH on oma väärtuselt
võrdne
pKa väärtusele või on sellele lähedal, siis muutub lahuse pH väikese koguse happe või
aluse lisamisel küllaltki vähe. Kõige vähem muutub lahuse pH väikese koguse happe või aluse
lisamisel kui lahuse
pH = pKa. Eeltoodule rajanebki nõrga happe-konjugeeritud alus lahuste
puhverdusvõime. Lahuse
puhverdusvõime tähendab, et lahuse pH püsib väikese koguse happe või
aluse lisamise tagajärjel ligikaudu muutumatuna. Puhverlahused on biokeemia praktikas
igapäevased töölahused ja ka valdav osa looduses toimuvast biokeemiast toimub puhverlahustes.
Oletame, et me tahame uurida mingit biokeemilist reaktsiooni pH = 4,00 juures. Oletame
veel, et reaktsiooni käigus, kas eraldub või liitub prootoneid ja me tahame vältida
reaktsioonikeskkonna pH olulist muutumist reaktsiooni käigus. Selleks peame me reaktsiooni läbi
viima nõrga happe ja tema konjugeeritud aluse lahuses ehk puhverlahuses. Meie katse puhul on
sobivaimaks sipelghappe baasil valmistatud
puhver , kuna sipelghappe
pKa väärtus (
pKa = 3,75)
on meie soovitud pH väärtusele kõige lähemal. Loomulikult tuleb puhvri
valikul silmas pidada, et
puhvri komponendid ei reageeriks meie poolt uuritava reaktsiooni
komponentidega või ei segaks
reaktsiooni mingil muul viisil. Sipelghappe konjugeeritud aluse (formiaat ioon) ja happe suhte
puhverlahuses leiame Henderson-Hasselbalchi võrrandist:
4,00 = 3,75 + log([HCOO-]/[HCOOH]) (3.16)
Avaldades siit otsitava suhte saame:
([HCOO-]/[HCOOH]) = 10pH-pKa = 100,25 = 1,78 (3.17)
Sellise puhverlahuse võib valmistada segades omavahel õigetes vahekordades kokku sipelghappe
lahust ja sipelghappe soola (näit. naatrium formiaadi) lahust. Kõige lihtsam viis on aga
valmistada sipelghappe lahus ja tiitrida see näiteks naatrium hüdroksiidiga kuni pH = 4,00. Lisaks
pH-le on puhverlahuste puhul oluliseks näitajaks veel
puhvermahtuvus . Mida suurem on
puhvermahtuvus, seda vähem muutub puhvri pH teatud koguse happe või aluse lisamisel.
Puhvermahtuvus on seda suurem, mida kõrgem on puhvri komponentide kontsentratsioon. Kui
suhe ([A-]/[AH]) on määratud soovitud puhverlahuse pH poolt, siis puhverdavate osakeste
üldkontsentratsioon, [A-] + [AH] on varieeritav.
Kuna biokeemilised reaktsioonid toimuvad enamasti füsioloogilise pH piirkonnas (pH 6,5
– 8,0), siis pakuvad biokeemikutele erilist huvi just sellesse piirkonda jäävate
pKa väärtustega
nõrgad happed. Tabelis 3.3 toodud hapetest
sobiksid füsioloogilise pH vahemikus kasutamiseks
divesinikfosfaat (H
2PO4 ) ja süsihape. Fosfaatpuhvrid on biokeemilises praktikas laialt levinud
8
kuid nad ei ole alati rakendatavad, kuna paljud biokeemilised reaktsioonid toimuvad fosfaatiooni
osavõtul. Lisaks võivad
fosfaat ja karbonaatpuhvrid sadestada mõningaid reaktsiooni seisukohast
vajalikke ioone (näit Ca2+). Tabelis 3.4 on toodud rida füsioloogilise pH piirkonnas kasutatavaid
looduslikke ja sünteetilisi puhvreid.
Ka organismid peavad säilitama nii oma rakkude sisekeskkonna kui ka kehavedelike pH
kitsas vahemikus (reeglina 6,5 – 8,0). Kuna fosfaatioon esineb rakkudes küllalt kõrgel
kontsentratsioonil ja divesinikfosfaadi (H
2-
2PO4 )
pKa = 6,86, siis on just
H2PO4 ↔ HPO4 + H+
tasakaal oluliseks puhversüsteemiks rakkude sisekeskkonnas. Kuna vere kaudu toimub
metabolismi jääkproduktina tekkinud CO2 eemaldamine, siis on vere pH määramisel olulisel
kohal karbonaatpuhver (
pKa = 6,37). Tulenevalt lahustunud süsihappegaasi reaktsioonist veega on
karbonaatpuhver tabelis 3.3 esitatust natuke keerulisem:
CO2 + H2O ↔ H2CO3 (3.18)
Lisaks neile ja
paljudele teistele madalmolekulaarsetest komponentidest koosnevatele puhvritele
mängivad ka osad makromolekulid (näit.
valgud ) raku pH kujundamisel olulist rolli. Valgud
sisaldavad oma koostises hulgaliselt erinevaid ioniseeritavaid gruppe, millest osade
pKa väärtused
langevad füsioloogilise pH vahemikku. Kuna valgud esinevad rakus ja ka kehavedelikes (näiteks
veri ja lümf) suhteliselt kõrgetes kontsentratsioonides ja lisaks sisaldab üks valgumolekul reeglina
terve rea ioniseeritavaid gruppe, siis on ka valkudel oluline osa bioloogiliste lahuste
puhvermahtuvuse määramisel.
Mitut ioniseeritavat gruppi sisaldavad molekulid: amfolüüdid, polüamfolüüdid ja
polüelektrolüüdid
Senini keskendusime peamiselt ainult ühte nõrgalt happelist või aluselist rühma
sisaldavatele molekulidele. Paljud molekulid sisaldavad aga mitut ioniseeritavat rühma ja omavad
seetõttu ka keerukamaid tiitrimiskõveraid.
Molekuli, mis sisaldab samaaegselt nii happelise kui aluselise
pKa väärtusega
ioniseeritavat rühma nimetatakse
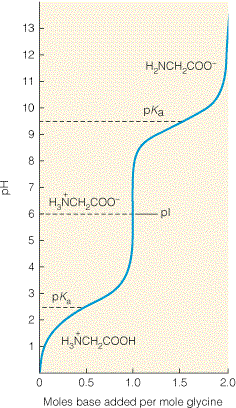
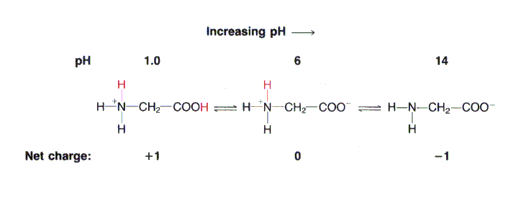
amfolüüdiks. Vaatame näiteks aminohapet glütsiin, H2N-CH2-
COOH. Glütsiini karboksüülrühma ja aminorühma
pKa väärtused on vastavalt 2,3 ja 9,6. Kui me
lahustame glütsiini väga happelises keskkonnas (näit. pH = 1,0), siis on nii glütsiini
karboksüülrühm kui aminorühm protoneeritud ja
molekul omab summaarset laengut +1. Hakates
nüüd lahuse pH-d tõstama (näit NaOH lisamise teel) hakkab toimuma prootoni dissotsiatsioon
vastavalt järgmisele skeemile:
Seega toimub glütsiini tiitrimine kahes
astmes . Kõigepealt loovutab prootoni karboksüülrühm ja
alles seejärel vähem happeline aminorühm (rääkides aminorühma –NH2
pKa väärtusest mõtleme
vastava konjugeeritud happe –NH +
3
pKa väärtust). Joonisel 3.7 toodud glütsiini tiitrimiskõverast
nähtub, et glütsiin võib toimida hea puhvrina kahes küllaltki
erinevas pH piirkonnas. Mõlemas
9
pH piirkonnas võime kasutada tiitrimiskõvera kirjeldamiseks vastava ioniseeritava grupi kohta
kehtivat Henderson-Hasselbalchi võrrandit.
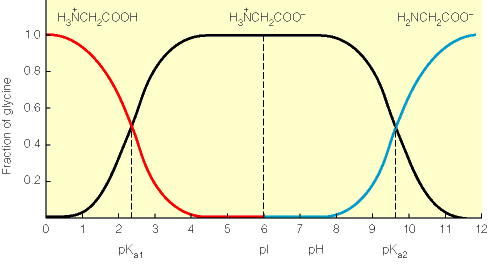
Madalatel pH väärtustel
domineerib glütsiini molekuli +1 laenguga vorm ja kõrgetel pH
väärtustel -1 laenguga vorm. Glütsiini erinevate ioonsete vormide suhteline sisaldus sõltuvalt
lahuse pH-st on toodud joonisel 3.8. Eriti huvipakkuv on olukord neutraalse pH piirkonnas.
Nimelt on selles piirkonnas enamus glütsiinimolekule vormis H3N+-CH2-COO- ja
molekulil puudub summaarne laeng. Sellises olekus, kus negatiivsete laengute hulk võrdub positiivsete
laengute
hulgaga , esinevat amfolüüti nimetatakse
zwitteriooniks.
Rangelt võttes esineb ainult üks
konkreetne pH väärtus, mille juures glütsiini
keskmine (kõigi lahuses esinevate
glütsiinimolekulide keskmine) laeng on null. Sellist pH väärtust nimetatakse
isoelektriliseks
punktiks,
pI. Kui pH =
pI, siis on valdav osa glütsiinimolekule kujul H3N+-CH2-COO- ning
vormid H3N+-CH2-COOH ja H2N-CH2-COO- esinevad väga väikeses kuid omavahel täpselt
võrdses hulgas. Me võime leida isoelektrilise punkti rakendades Henderson-Hasselbalchi
võrrandit kummagi ioontasakaalu jaoks eraldi:
pI =
pKCOOH + log([H3N+CH2COO-]/[H3N+CH2COOH]) (3.19)
ja
pI =
pKNH3+ + log([H2NCH2COO-]/[H3N+CH2COO-]) (3.20)
liites võrrandid 3.19 ja 3.20 ning koondades logaritmitavad suurused ühe logaritmimärgi alla
saame:
2pI =
pKCOOH +
pKNH3+ + log([H2NCH2COO-]/[H3N+CH2COOH]) (3.21)
Kuna isoelektrilises punktis on [H2NCH2COO-] = [H3N+CH2COOH], siis saame:
pI = (pKCOOH + pKNH3+)/2 (3.22)
Seega näeme, et kahte ioniseeritavat rühmi sisaldava molekuli
pI on lihtsalt vastavate rühmide
pKa väärtuste aritmeetiline keskmine. Glütsiini
pI on seega
pI = (2,3 + 9,6)/2 = 5,95. Nagu me
jooniselt 3.8 näeme, on glütsiin valdavalt zwitterioonses vormis pH vahemikus ligikaudu 4 – 8.
Suured molekulid nagu valgud võivad sisaldada palju happelisi ja aluselisi rühmi.
Selliseid molekule nimetatakse
polüamfolüütideks. Juhul kui molekul sisaldab rohkem kui kaks
ioniseeritavat rühma läheb
pI arvutus keeruliseks. Siiski võib alati öelda, et kui molekulis on
esindatud nii positiivselt kui negatiivselt laetud rühmad, eksisteerib ka pH väärtus, mille juures
molekulide keskmine laeng on null ehk molekulil esineb isoelektriline punkt. Kui molekulis
domineerivad
happelised rühmad on
pI madal, kui aga domineerivad aluselised rühmad on
pI kõrge. Konkreetse valgu
pI väärtus on seda valku iseloomustavaks suuruseks.
Amfolüütide ja polüamfolüütide
pI on võimalik määrata eksperimentaalselt. Selleks
kasutatakse elektroforeesi pH gradiendis ja seda nimetatakse
isoelektriliseks fokuseerimiseks .
Elektrivälja toimel amfolüütide lahusele hakkavad summaarset
positiivset laengut
kandvad molekulid liikuma katoodile (katioonid liiguvad katoodile) ja negatiivset kogulaengut kandvad
molekulid liiguvad anoodi suunas (
anioonid liiguvad anoodile). Isoelektrilises punktis on
molekulide keskmine laeng null ja seetõttu nad elektriväljas ei liigu. Kuna
elektroforees viiakse
läbi pH gradiendis siis liiguvad amfolüüdid, seni kuni satuvad geelis kohta, kus
pH = pI ja sellest
punktist nad edasi ei liigu. Lisaks
pI määramisele saab isoelektrilise fokuseerimise abil lahutada
ka erinevaid amfolüüte nende segudest.
Mõned makromolekulid sisaldavad suurel hulgal kas ainult positiivseid või ainult
negatiivseid laenguid. Selliseid molekule nimetatakse
polüelektrolüütideks. Tugevad
10
polüelektrolüüdid, nagu negatiivselt laetud
nukleiinhapped , on ioniseeritud laias pH vahemikus.
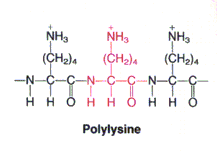
Esinevad ka nõrgad polüelektrolüüdid nagu polülüsiin (aminohappe lüsiin homoploümeer):
Kui ühe molekuli koostises on terve rida nõrku ioniseeritavaid rühmi, siis võib mingi
ioniseeritava rühma
pKa väärtus sõltuda naaberrühmade ioniseerituse olekust. Näiteks polülüsiin
loovutab oma esimesed
prootonid tänu positiivsete laengute tõukumisele palju kergemini kui
viimased. Polülüsiinis on protoneeritud rühmad positiivselt laetud ning tõukuvad, prootoni
eemaldamisega väheneb ka energeetiliselt ebasoodne tõukejõud (esimesed prootonid tõugatakse
eemale ja nende
pKa on vastava mõjutamata grupi
pKa-st väiksem). Mõneti sarnane on olukord ka
juhul kui prootoni loovutamisega kaasneb negatiivse laengu teke. Kui esimesed prootonid
eemaldatakse vastavalt konkreetsete gruppide
pKa väärtustele, siis viimaste gruppide
deprotoneerimine on raskendatud negatiivsete laengute tõukumise tõttu (viimaste prootonite
eemaldamise
pKa on esimeste, mõjutamata gruppide
pKa -st oluliselt suurem).
Makroioonide vahelised interaktsioonid lahuses
Suured polüelektrolüüdid nagu nukleiinhapped ja polüamfolüüdid nagu valgud
koondatakse ühise nimetuse
makroioonid alla. Sõltuvalt keskkonna pH-st võib makroioonidel
esineda märkimisväärne summaarne laeng. Makroioonide vahelised elektrostaatilised tõmbe- ja
tõukejõud mängivad olulist rolli nende käitumise määramisel vesilahuses.
Makroioonide lahustuvus ja pH
Kuna samamärgilist kogulaengut kandvad makroioonid tõukuvad omavahel, siis omavad
nukleiinhappe molekulid tendentsi hoiduda lahuses teineteisest eemale (joonis 3.9a). Samal
põhjusel on ka valkude lahustuvus suurem pH väärtustel üle või alla valgu
pI väärtust, kus
valgumolekulidel esineb summaarne laeng. Erimärgilisi summaarseid laenguid kandvate
makroioonide vahel esineb aga elektrostaatiline tõmbumine ja sellised molekulid omavad
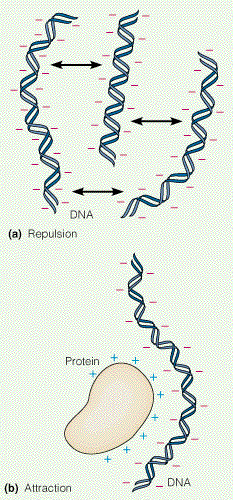
tendentsi agregeeruda (joonis 3.9b). Paljud valgud interakteeruvad tugevasti DNA-ga ja sellistele
valkudele on iseloomulik tugeva positiivse laengu esinemine (joonis 3.9b). Näitena võib siin tuua
kõrgemate organismide kromosoomide organisatsiooni. Negatiivselt laetud DNA on tugevalt
assotsieerunud positiivselt laetud valkude
histoonidega moodustades kompleksi, mida
nimetatakse kromatiiniks.
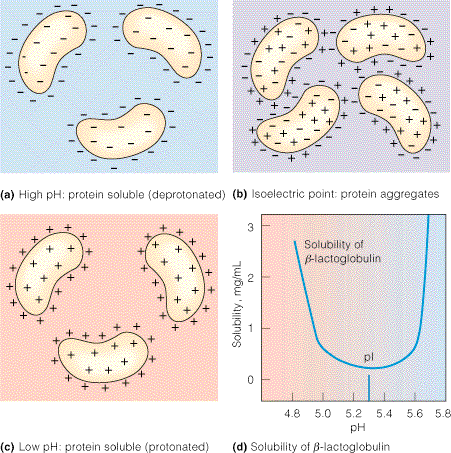
Mõningatel valkudel on kalduvus omavahel agregeeruda kui pH on võrdne nende
pI-ga
(joonis 3.10). Näiteks tavaline piimas esinev valk β-laktoglobuliin on polüamfolüüt
pI väärtusega
5,3. Kui pH on sellest väärtusest suurem või väiksem, siis omavad kõik molekulid kas
summaarset negatiivset või positiivset laengut ja tõukuvad omavahel (joonis 3.10a,c). Seetõttu on
selle valgu lahustuvus nii aluselises kui happelises keskkonnas väga kõrge (joonis 3.10d). Kui pH
on võrdne isoelektrilise
punktiga , siis on valgumolekulide keskmine laeng null. Sellele vaatamata
sisaldavad valgumolekulid oma pinnal erinevat laengut kandvaid piirkondi. Erimärgilise laenguga
piirkondade omavaheline tõmbumine koos van der Waalsi jõududega põhjustab valgumolekulide
agregeerumist ja lahusest välja sadenemist (joonis 3.10b). Seetõttu on β-laktoglobuliini nagu ka
paljude teiste valkude lahustuvus isoelektrilises punktis minimaalne (joonis 3.10d).
11
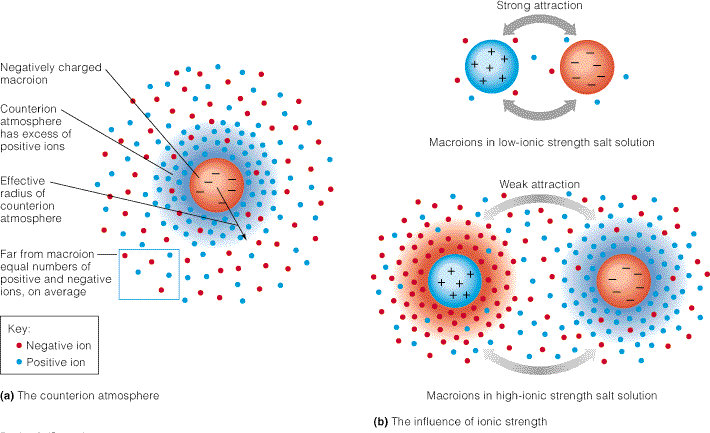
Väikeste ioonide mõju: ioonne jõud
Makroioonide omavaheline interaktsioon on tugevasti mõjutatud väikeste ioonide, nagu
lahustunud
soolad , olemasolu poolt. Iga makroioon koondab enda ümber väikestest
vastasioonidest
koosneva ioonatmosfääri. See erimärgilistest ioonidest koosnev pilv varjestab
makroioonide laenguid ja takistab neil seeläbi omavahel interakteerumast (joonis 3.11a). Kuigi
varjestusefekti sõltuvus väikeste ioonide kontsentratsioonist on keeruline, on ilmne, et mida
kõrgem on väikeste ioonide kontsentratsioon, seda tugevam on nende poolt avaldatav
makroioonide laenguid varjestav toime. P. Debye ja E. Hückel kasutasid mõistet ioonatmosfääri
efektiivne raadius (
r) kirjeldamaks väikeste ioonide mõju sfääriliste makroioonide omavahelisele
interaktsioonile. Ioonatmosfääri efektiivset raadiust võib vaadelda kui
vahemaad , mille tagant
kaks makroiooni teineteise kohalolekut veel „tunnetavad”. Vastavalt Debye-Hückeli
teooriale :
r = K/√I (3.23)
kus
K on konstant, mis sõltub keskkonna dielektrilisest konstandist ja temperatuurist ning
I on
lahuse
ioonne jõud.
I sõltub lahustunud ioonide kontsentratsioonist
M ja laengust
Z:
I = ½∑MiZ2i (3.24) Seega on ioonne jõud summa, mis
arvestab kõikide väikeste ioonide kontsentratsioone ja
laenguid. 1:1 elektrolüüdi nagu NaCl puhul on
ZNa+ = +1 ja
ZCl- = -1 ja kuna
MNa+ =
MCl- =
MNaCl , siis on NaCl lahuse ioonne jõud arvuliselt võrdne tema molaarse kontsentratsiooniga. See
kehtib kõikide 1:1 elektrolüütide puhul kuid ei pea paika multivalentsete ioonide (Mg2+ või SO 2-
4 )
esinemise korral. Kuna ioonse jõu avaldises (3.24) on iooni laeng
ruudus , siis omavad
multivalentsed ioonid ioonatmosfääris suuremat individuaalset mõju kui monovalentsed ioonid.
Selliste elektrolüütide puhul on ioonne jõud suurem nende molaarsest kontsentratsioonist.
Keskkonna ioonse jõu mõju makroioonide vahelisele interaktsioonile võib lihtsustatult
kokku võtta vastavalt joonisele 3.11b. Madala ioonse jõu korral on vastasioonidest koosnev
ioonatmosfäär hõre ning hajus. Ioonatmosfääri varjestav efekt on väike ja makroioonide vahel
valitseb tugev elektrostaatiline interaktsioon. Ioonse jõu kasvades muutub makroioone ümbritsev
ioonatmosfäär tihedamaks ja kontsentreeritumaks ning omab tugevat varjestavat efekti. Seetõttu
ongi makroioonide vahelised elektrostaatilised interaktsioonid kõrge kontsentratsiooniga
elektrolüütide (soola) lahuses nõrgad.
Ioonatmosfääri varjestav efekt aitab seletada üldist tähelepanekut, et lahuse ioonse jõu
suurendamine (teatud piirini) suurendab valkude lahustuvust ja seda isegi isoelektrilise punkti
juures. Seda efekti nimetatakse valkude sisse-soolamiseks (ingl. salting in). Tõstes ioonset jõudu
veelgi (näiteks kuni mõne molaarse soola kontsentratsioonini) hakkab toimuma
vastupidine efekt,
valkude lahustuvus väheneb. See on
seletatav vee aktiivsuse vähenemisega. Väga
kontsentreeritud soolalahustes on enamik veemolekule, mis muidu osalesid valkude
solvatatsioonil (olid seotud valgumolekulidega) kaasatud soolaioonide hüdratatsioonikihtidesse.
Valgumolekulide lahustumiseks ei jätku lihtsalt piisavalt vabasid veemolekule. Valkude
lahustuvuse vähenemist väga kõrgetel soola kontsentratsioonidel nimetatakse valkude välja-
soolamiseks (ingl. salting out). Kuna erinevad valgud käituvad eeltoodu suhtes pisut erinevalt,
siis võib nii valkude sisse- kui välja-soolamist kasutada valkude eraldamisel nende segudest.
Kuna lisaks pH-le on makroioonide interaktsioonid mõjutatud ka ioonsest jõust siis peab
biokeemilises praktikas lisaks pH kontrollimisele puhverlahuste kasutamisega kontrollima ka
ioonset jõudud. Ioonset jõudu kontrollitakse enamasti neutraalsete (pH mõttes) soolade nagu
NaCl ja KCl abil. Kuigi erinevate rakkude ja kehavedelike ioonne jõud võib olla küllaltki erinev,
on biokeemilises praktikas enamasti sobilik 0,1 – 0,2M NaCl-le vastav ioonne jõud.
12
Kokkuvõte
Vesi on elu eksisteerimise seisukohast asendamatu keskkond. Enamik vee unikaalseid
omadusi tuleneb veemolekulide polaarsusest ja võimest moodustada omavahel vesiniksidemeid.
Vesi on suurepärane solvent. Polaarsed, vesiniksidemeid moodustavad ja
ioonsed ühendid
lahustuvad vees hästi ja neid nimetatakse hüdrofiilseteks. Hüdrofoobsed ühendid lahustuvad vees
ainult piiratud
kogustes . Amfipaatsed molekulid koosnevad hüdrofiilsest ja hüdrofoobsest osast.
Veega kontaktis olles moodustavad amfipaatsed molekulid erinevaid struktuure nagu
monomolekulaarsed kihid, vesiikulid ja mitsellid. Sellised molekulid moodustavad ka
membraanseid kaksikkihte, mis ümbritsevad rakke ja organelle.
Nõrkade hapete ja aluste ionisatsioonil on biokeemias äärmiselt oluline roll. Enamik
biokeemilisi protsesse toimub pH vahemikus 6,5 – 8,0 ja seda nimetatakse füsioloogiliseks pH
vahemikuks. Nõrga happe ja vastava konjugeeritud aluse käitumine on kirjeldatav Henderson-
Hasselbalchi võrrandiga, mis seob omavahel [konjugeeritud alus]/[dissotsieerumata hape] suhte,
pH ning
pKa. Nõrga happe tiitrimiskõverad näitavad, et nõrga happe lahuse pH muutub happe või
aluse lisamisel kõige vähem siis, kui pH on võrdne nõrga happe
pKa-ga ja sellel põhineb
puhverlahuste valmistamine.
Amfolüüt on molekul, mis sisaldab nii happelist kui aluselist ioniseeritavat rühma.
Sõltuvalt keskkonna pH-st võib amfolüüdi summaarne laeng olla kas positiivne, null või
negatiivne. Polüamfolüüt sisaldab palju happelisi või aluselisi rühmi. Amfolüüdi või
polüamfolüüdi isoelektriline punkt on pH väärtus, mille juures amfolüüdi või polüamfolüüdi
molekulide keskmine summaarne laeng on null. Polüelektrolüüdid on molekulid, mis kannavad
paljusid samamärgilisi laenguid. Makroioonide (polüamfolüüdid, polüelektrolüüdid) käitumine
sõltub lahuse pH-st ja väikeste ioonide olemasolust lahuses. Väikesed ioonid varjestavad
makroioonide laenguid ja nõrgestavad makroioonide vahelisi interaktsioone. Varjestamise ulatus
sõltub lahuse ioonsest jõust ja seda kirjeldab kvantitatiivselt Debye-Hückeli teooria.
13
Loeng III joonised
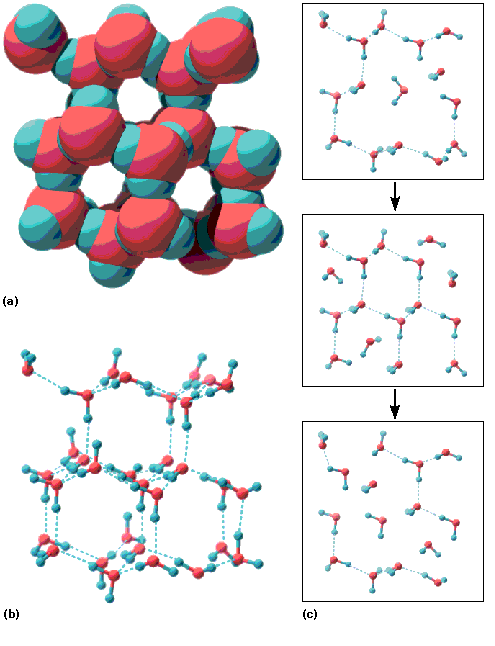
Tabel 3.1. Vee omadused võrrelduna mõnede madalmolekulaarsete ühendite omadustega. Tabel 3.2. Mõningad vee omadused võrrelduna vesiniksidemeid mittemoodustava ja mittepolaarse ühendi, n-pentaaniga. Joonis 3.1. Vesiniksidemete
moodustumine vees. a)
veemolekuli elektronstruktuur,
vabad elektronpaarid on headeks
vesiniksideme aktseptoriteks. b)
Vesiniksidemete moodustumine
veemolekulide vahel.
Veemolekul võib samaaegselt
olla nii vesiniksideme doonoriks
kui aktseptoriks.
14
Joonis 3.2. Jää molekulaarne võre. a) Jää ruumiline mudel. Jää on omavahel
vesiniksidemetega ühendatud veemolekulidest koosnev korrapärane struktuur. Kuna
veemolekul saab olla korraga kahe vesiniksideme aktseptoriks ja kahe vesiniksideme
doonoriks, siis moodustub korrapärane tetraeedriline struktuur, kus iga veemolekul on
ühendatud nelja naaber veemolekuliga. b) Jää
skelett -mudel. c) Vee struktuur vedelas olekus.
Jää sulades laguneb suurem osa korrapärasest struktuurist kuid mingid jää struktuuri
fragmendid säilivad ka vedelas olekus. Vee struktuuri moodustavad n.ö. vahelduvad klastrid,
mis koosnevad teineteisega vesiniksidemeta kaudu seotod teatud hulgast veemolekulidest.
Klastreid koos hoidvad vesiniksidemed on pidevas ja kiires katkemise ja reformeerumise
protsessis. Kolm järjestikust “pilti” on võetud 1 pikosekundiliste (1 ps = 10-12s)
ajavahemike järel.
15
Joonis 3.3. Ioonide hüdratatsioon vesilahuses. Ioonsed ühendid nagu NaCl
lahustuvad vees hästi, kuna vesilahuses moodustub ioonide ümber
veemolekulidest hüdratatsiooni kiht.
Joonis 3.4. Amfipaatsed molekulid. Amfipaatne molekul koosneb
hüdrofiilsest osast (pea) ja hüdrofoobsest osast (saba).
16
Tabel 3.3. Mõned nõrgad happed koos vastavate konjugeeritud alustega. Tabel 3.4. Mõned biokeemilises praktikas sagedasti
kasutatavad puhvrid . Joonis 3.5. Mõnede kehavedelike ja
tarbekaupade pH väärtused. 17
Joonis 3.7. Amfolüüt glütsiini Joonis 3.6. Nõrkade hapete tiitrimiskõver. Kuna glütsiin sisaldab
tiirimiskõverad. Joonisel on toodud kahe
kahte ioniseeritavat rühma, mille
pKa nõrga happe, sipelghappe (HCOOH) ja
väärtused on teineteisest piisavalt
ammooniumiooni (NH4 ) tiitrimiskõverad.
erinevad (vastavalt 2,3 ja 9,6), siis on
X-teljel on algse happe moolide arvu
glütsiini tiitrimiskõver selgelt
suhtes lisatud aluse moolide arv. Pange
kaheastmeline. Joonisel on näidatud ka
tähele, et tiitrimiskõvera keskpunktis
glütsiini jaoks arvutatud isoelektriline
(lisatud on poolele algse happe hulgale
punkt
pI.
vastav hulk alust) on lahuse pH võrdne
nõrga happe
pKa väärtusega. Selles
piirkonnas (± 1 pH ühik) kulub lahuse pH
muutmiseks ka kõige suurem hulk alust.
Kõige rohkem alust kulub lahuse pH
muutmiseks mingi vahemiku võrra punktis,
kus pH =
pKa, järelikult on see antud happe
poolt kõige paremini puhverdatud piirkond.
18
Joonis 3.8. Glütsiini kolme erineva ioonse
vormi suhtelised hulgad sõltuvalt lahuse pH-
st. Terves füsioloogilise pH vahemikus on
glütsiin zwitterioonses vormis. Joonisel on
näidatud ka glütsiini karboksüülrühma ja
aminorühma
pKa väärtused ning molekuli
pI väärtus.
Joonis 3.9. Makroioonide vahelised
elektrostaatilised interaktsioonid. a)
Kuna DNA molekulid on füsioloogilisel
pH-l negatiivselt laetud, siis esineb lahuses
DNA molekulide vahel elektrostaatiline
tõukumine. b) Positiivselt laetud
valgumolekuli ja DNA molekuli vahel
toimub elektrostaatiline tõmbumine.
19
Joonis 3.10. Valgu lahustuvuse
sõltuvus pH-st. Enamik valke on
hästi lahustuvad kas kõrgel või
madalal pH-l, kus
valgumolekulidel on
samamärgiline laeng, mis väldib
valkude agregeerumist (a ja c).
Isoelektrilises punktis on
valgumolekulide summaarne
laeng küll null, kuid molekulid
sisaldavad nii positiivselt kui
negatiivselt laetud piirkondi, mis
omavahel tõmbudes põhjustavad
valkude agregeerumist (b). β-
laktoglobuliini lahustuvus on
minimaalne isoelektrilises
punktis.
Joonis 3.11. Väikeste ioonide mõju makroioonide vahelistele interaktsioonidele. a) Kui
laenguga makroioon asetada soola vesilahusesse, siis klasterduvad tema ümber väikesed
vastasmärgilised ioonid, moodustades vastaslaengutest ioonatmosfääri. b) Madala ioonse jõuga
lahustes on makroioonide vaheline interaktsioon tugevam kui kõrge ioonse jõuga lahuses, kus
makroioonide ümber moodustunud ioonatmosfäär varjestab nende laenguid.
20






































Kõik kommentaarid