Lora Sulg ,
Proviisor II, sügis 2010
1. OPTILISED MEETODID.
Optiliste meetodite korral kasutatakse aine võimet mõjutada valguskiirguse omadusi, nagu
intensiivsus, sagedus, levimiskiirus, polarisatsioonitasand.
Valguskiirgus- elektromagnetkiirguse diapasoon, kuhu kuuluvad ultravioletkiirgus (1-400nm), nähtav
kiirgus (400-800nm), infrapunakiirgus (800-1000000nm). Farmatseutilises analüüsis kasutatakse
kõige enam vahemikku 190-400 nm.
Valge värv on kogu spektri värvuste segu. Sinine, roheline ja punane on põhivärvused ja
nendest sünteesitakse kõik värvused. Purpurpunane ja taevassinine on täiendvärvid, millest
tinglikult sünteesitakse must värvus.
Mida väiksem
lainepikkus , seda rohkem energiat.
1.1 REFRAKTOMEETRIA.
Valguskiirguse levimise suuna muutumine ehk
murdumine ehk
refraktsioon on põhjustatud valguse
levimiskiiruse muutumisest üleminekul ühest keskkonnast teise. Selle
tingimuseks on keskkondade
erinev tihedus.
Mida tihedam on keskkond, seda aeglasemalt
elektromagnetkiirgus ehk valgus levib.
Vaakumis on see
300000 km/s. Õhus see kiirus langeb. Vees on see umbes 250000 240000 km/s.
Murdumisnäitaja võrdub lainete levimiskiiruse suhtega kahes keskkonnas. Tähistus n.
Kus n murdumisnäitaja alfa- langemisnrk
beta murdumisnurk v1 lainete levimise kiirus esimeses kkonnas v2 lainete levimise kiirus teises kkonnas
Valguse üleminekul tihedamast kkonnast hõredamasse on murdumisnurk beta lahgemisnurgast alfa
suurem, ülemiekul hõredamast tihedamasse väiksem.
Valguse murdumise intensiivsust mõõdetakse refraktomeetriga. Kasutatakse
kriitilise nurga
printsiibi (krit nurk nurk, millest alates toimub täielik
sisepeegeldus ). Esimene
prisma pind on
vedelikuga kokkupluutekohalt matistatud. Matistatud pind tekitab intensiivse sisepeegelduse, millest
tuleneb
okulaari vaateväljas tume ala, vedelikukihi läbinud refrakteerunud kiired tekitavad vaateväljas
heleda ala. Hele ja tume ala on eraldatud terava eraldusjoonega. Mida suurem on vedeliku
murdumisnäitaja, seda rohkem kallutatakse valguskiiri optilisest teljest kõrvale ja seda laiem on tume
ala.
Kuna murdumisnurk sõltub lainepikkusest, siis määramine toimub mingil kindlal lainepikkusel.
Murdumisnäitaja sõltub ka oluliselt temperatuurist. Kõik mõõtmistulemused on antud 20oC juures.
Vajadusel saab arvutada ka paranduskoeffitsent.
x=(n-n20)/(20-t).
kus x paranduskoeff n refraktomeeri näit n20 murdumisnäitaja 20C t uuritava lahuse t-tuur
Iga
refraktomeeter tuleb enne kasutamist kalibreerida e viia mõõtskaala näit vastavusse
murdumisnäitaja tegeliku väärtusega.. Selleks kasutatakse vett. Vee murdumisnäitaja peab olema
vähemalt 1,333 õhu suhtes.
Miinuseks on väike tundlikkus ja valimatus. Kahe aine segus ühte kindlat ainet ei ole võimalik määrata.
Plussiks on ääretu lihtsus.
Piisab väga väikesest ainekogusest (tilgake). Sellega saab ka määrata kõiki
aineid, mis pole optiliselt aktiivsed, pole redoksomad-ga, ei ole värvilised ega oma UV-
alas iseloomusikke neeldumimax-me, nagu pasljud
suhkrud (
sahharoos ) Lora Sulg, Proviisor II, sügis 2010
1.2 POLARIMEETRIA. Polarimeetria füüsikaline alus on ainete optiline aktiivsus. Optiline aktiivsus ehk optiline pöörang on aine võime pöörata seda läbiva
polariseeritud valguse polarisatsioonitasapinda. Kui polarisatsioonitasand pöördub
kellaosuti liikumise suunas,
tähistatakse seda ,,+" märgiga (paremalepöörav, D) ja kui kellaosuti liikumise
vastassuunas , siis ,,-"
märgiga (vasakulepöörav, L). Polarisatsioonitasandi kõrvalekalde nurka algtasemest nimetatakse pöördenurgaks ja seda
tähistatakse -ga. Pöördenurga suurus sõltub 1) optiliselt aktiivse aine omadustest ehk aine
aktiivsusest, 2) polariseeritud valguse lainepikkusest, 3) teepikkusest (mida pikem tee, seda suurem on
pöördenurk) ja 4) lahustist teatud määral. Enamasti tegu vesilahusega.
Kuna erinevatel
ainetel on erinevad omadused, siis ühtlustamiseks on sisse toodud
eripöörangu mõiste ([]). Eripöörang näitab, kui aktiivselt, kui tugevalt muudab antud aine
polarisatsioonitasapinda. Eripöörang on arvutuslik suurus, mis väljendab monokroomse valguse
pöördenurga muutust, kui see on läbinud teepikkuse 1 dm keskkonnas (s.o. lahusekihi paksus), mis
sisaldab optiliselt aktiivset ainet kontsentratsioonis 1 g/ml. Mida suurem eripöörang, seda optiliselt
aktiivsem on aine. Eripöörang alati määratakse ja kõik pöördenurga muutused määratakse temperatuuril 20oC ja
lainepikkusel (Na-lainepikkus) 589,3 nm. Alati märgitakse ära ka lahusti
[] = (a*100)/(l*c), kus alfa- mõõdetud pöördenurk l lahusekihi paksus (dm) c lahuse kontsentr. (gr/100ml lahuse)
vedelike jaoks : [] = /l*p kus p vedeliku tihedus (gr/1ml)
kontsentratsiooni leidmine: c = ( *100)/([]*l)
Valgus, mis näiteks päikeselt tuleb, on polariseerimata. Valgus võngub täiesti suvalistes tasandites. Kui
panna ette spetsiaalse
filtri , mis
laseb valgust läbi ainult teatud suunas, siis kogu valgusvihust tuleb läbi
ainult see valgus, mis võngub filtri tasapinnaga samas suunas. Selline valgus on juba polariseeritud.
Veepinnalt peegeldudes valgus osaliselt polariseerub. Kõik vedelkristalldispleid (nt. käekell) töötavad
polarifiltritel. Risti polarisatsioonitasand on tume, välja lülitades ei ole näha.
Uuritava aine konts saab määrata polarimeetriga. See koosneb monokromaatse valguse allikast
(naatriumlamp või hõõglamp kollase filtriga), polaristasioonifiltrist, küvetist uuritava lahusega, teisest
polar.
filtrist ning vaatlusokulaarist.
Valgusallikast lähtua monokromaatse valgusekiirguse polariseerib, esimene polarifilter. Peale küvetti
asuv teine polarifilter keeratakse esimese suhtes 90 kraadi nurga alla ehk risti. Püütakse saavutada
olukord, kus läbi kahe filtri läbitakse võimalikult vähe valgust.
ÜLESANNE. Sahharoosi lahus. Eripöörang on +66,65oC. Küvett 20 cm pikk. Pöördenurk oli 12,57o.
Protsendiline kontsentratsioon?
c = ( *100)/([]*l)
c=(12,57*100)/(66,65*2)=9,43% Lora Sulg, Proviisor II, sügis 2010
1.3. SPEKTROFOTOMEETRIA.
Spektrofotomeetriat kasutatakse individuaalsete ainete ja
segude kvalitatiivseks ja kvantitatiivseks
analüüsiks.
Reeglina sobib puhaste ainete analüüsil. Teatud juhtudel võib analüüsida ka 2-st kuni 3-st
komponendist
koosnevat segu. Kvalitatiivne analüüs on raskendatud, kuid kvantitatiivset saab teostada.
Vahendiks on spektrofotometer, mis võimaldab mõõta valguse neeldumise intensiivsust proovis. Kuna
neeldumisintensiivsus sõltub valguse lainepikkusest, peab spektrofotomeeter võimaldama valida
spektrista antud ühendi jaoks sobiva lainepikkuse.
Nähtavale spektriosale vastavad lainepikkused 380-780 nm, saab mõõta vaid värviliste lahuste
opt.
tihedust .
UV alas (185-380nm)
neelavad valgust praktiliselt kõik orgaanilised ained. Spetsiifilisem on
neeldumine 280-380 nm, seda laineala kasut aine identifitserimiseks neeldumismaksimume järgi.
Spektrofotomeeter.
Valgusallikas
halogeenlamp või deuteeriumlamp.
Monokromaator
difraktsioonivõre või kolmnurkne prisma, polükromaatne valgus lahutatakse peegeldumisel spektriks.
Pilu - mõõtmed määravad kui lai spektriosa läbib uuritava proovi. Mida kitsam pilu, seda täpsem
spekter registreeritakse. Suurendades pilu, seda suurem tundlikkus (seda väiksem kontsentratsioon on
võimalik määrata). Valgustundlik element fotoelektronkordisti, mõõdab küveti läbinud valguse
intensiivsust.
Skaneeriv spektrofotomeeter laseb korraga läbi pilu terve spektri. Jadadioodspektrofotomeetris
lahutatakse polükroomne valgus spektriks peale proovi läbimist ja kogu spekter suunatakse korraga
dioodide jadale. Nii registreeritakse reaalajas valguse neeldumine kogu spektrialas mitte vaid ühel
lainepikkusel.
Ühekanalisel spektrofotomeetril tuleb kordamööda liigutada prooviga küvett ja võrdlusküvett
valgusvihu ette. Kahekanaline toimub võrdlus korraga prooviküveti suhtes. Kõigepealt nullistatakse
viga küvetimaterjali suhtes ja mõõtes saame kohe võrdluse. Kahekanaline on kallim.
Fotoelektrokolorimeetris ei kasutata monokromaatorit vaid polükromaatset valgusest eraldatakse
kindel spektriosa filtre või värvilisi valgusdioodide abil. Kasutatakse ainult nähtavat spektriosa, st saab
mõõta ainult värviliste lahuste opt.tihedust.
Küveti materjal sõltub
kasutatava valsude lainepikkusest. Nähtavas alas sobib klaasist küvetid, UV
alas kvartsküvetid, kuna taaline klaas on UV-kiirguse jaoks läbipaistmatu. Enimlevinud läbimõõt on
10mm.
Lambert-
Beer 'i seadus.
Lahuse optiline tihedus on võrdelises sõltuvuses aine kontsentratsioonist lahuses. D või A
(absorbance).
Io proovile langenud kiirguse intensiivsus
I proovi läbinud kiirguse intensiivsus
Ehk siis mida rohkem läbis, seda vähem neeldus. Logaritmi kasutatakse mugavuse huvides.
Kui proovi läbis täpselt sama palju, kui proovile langes. Seega D=0.
Kui I0=1 I=0,5 D=0,301
Kui Io=1 I=0,1 D=1
Kui Io=1 I=0,01 D=2
Kui Io=1 I=0,001 D=3
D=xcb
x erineeldumisnäitada (aine võime neelata elektromagnetkiirgust)
c kontsentratsioon
b küveti läbimõõt (n.ö.
teepikkus , mida valgus peab läbima)
kontsentratsiooni leidmine: c = D/xb
Neeldumisnäitaja on füüsikaline
komponent , mis väljendab aine neelamisvõimet. Erinevatel
lainepikkustel on aine neelamisvõime erinev. Lora Sulg, Proviisor II, sügis 2010
Neeldumisnäitaja sõltub:
1) lahusekihi paksus (e. optilise tee pikkus)
2) lahuse
konsentratsioon (mida kontsentreeritum, seda rohkem neelab)
3) lainepikkus (erinevad ained neelavad kiirgust erinevatel lainepikkustel)
4) aine keemiline ehitus (kuidas on paigutunud ja millised need
aatomid on)
Erineeldumisnäitaja (x, A1%1cm) näitab kui suur oleks tinglikult optiline tihedus, kui lahuse konts oleks
1% ja lahusekihipaksus 1cm. .
Molaarne neeldumisnäitaja (epsilon) kui suur oleks lahuse opt.tihedusm kui lahuse konts oleks 1
mool/l ja lahusekihi paksus 1cm.
kus M on
molaarmass .
Optiline läbitavus e läbilaskvus (T) näitab mitu protsenti lahusele
langenud valgusest läbis selle.
Kuna lahuse optiline tihedus on linearses sõltuvuses lahuse kontentratsiooniga vaid teatud
kontsentratsioonivahemikus, on kasutatavad kontsentratsioonid on suhteliselt madalad. Suuremate
kontsentr korral
tekkivad protsessid nagu
dissotsiatsioon ja kompleksimoodustamine. Seetõttu
koostakse enne mõõtmist kalinratsioonigraafik, kust eitakse lahuse optilisele
tihedusele vastav
kontsentratsioon.
Valitakse selline lainepikkus, mida uuritava aine lahus kõige rohkem neelab ning lahuse kontsentr ja
kihi paksus selline, et mõõdetav opt.tihedus jääks
piiridesse 0,2-0,9 ühikut. Lahusti enese opt.tihedus
ei tohiks ületada 0,2 ühikut. Tavaliselt aine konts on 0,01-0,001%.
Kalibratsioonigraafik.
y=mx+b
m tangens sirge tõusunurgast e. sirge tõus
b sirge lõikepunkt y-
teljega selle abil saab leida kontsentratsoooni sõltuvalt optilisest tihedusest.
ÜLESANDED
1.Milline oli proovi optiline tihedus, kui võrdluslahusega küveti korral oli fotoelemendi pinge oli 4,5V ja
prooviküveti korral 2,3V?
D=log 4,5/2,3=0,291
Kasutatakse logaritmi, et suurt numbrite diapasooni võimalikult kompaktseks muuta.
2. Milline on aine protsentuaalne sisaldus, kui 230 nm juures 1% küvetis ja aine erineeldumisnäitaja on
480 (samadel tingimustel). Optiline tihedus on 0,700. Milline on aine kontsentratsioon?
c = 0,700/480 = 0,001458%
c = D/xb
3. Aine kontsentratsioon 0,001%, lainepikkusel 360 nm on optiline tihedus 0,480. Mis on selle aine
erineeldumisnäitaja?
0,001% 0,480
1% x
A1% = 480/0,001= 480 Lora Sulg, Proviisor II, sügis 2010
1.4 SPEKTROFOTOMEETRIA INFRAPUNAALAS. (IR)
Põhineb
infrapunakiirguse neeldumisel aines, kutsudes esile aatomivaheliste sidemete paindeid ja
vibratsioone. Kiirguse neeldumisel sidemed painduvad, nurgad sidemete vahel võivad muutuda.
Eripäraks on suurem infohulk ja see meetod annab infot molekuli struktuuri kohta.
Infrapunaspekter ei sõltu mitte ainult molekuli keemilisest struktuurist vaid ka aine
kristallmodifikatsioonist see võimaldab eristada ühe ja sama aine mitut polümorfset struktuuri.
IR-laineala on 800nm-1000 µm. Jaotatakse: lähi (0,8-02µm), kesk (2-15µm), kauginfrapunaspekter (15-
1000µm).
Analüüsis kasutatkse 4000-250 cm-1
Lainepikkuse mõõtmiseks kasutatkse pöördcm (cm-1), sest mikromeeter on suur õhik ja tuleb kasutada
komasid.
1000cm -1 = 1/1000cm ehk 0,001 cm = 0,01 mm = 10 µm = 10000 mm
2500nm (:1000) = 2,5µm (:1000) = 0,0025mm (:10) = 0,00025cm = 4000cm-1
Identifitseerimine toimub graafikute võrdlemise teel. X-teljele kantakse lainepikkus lainearvudes ja Y-
teljele proovi läbilaskvus protsentides (UV- ja nähtava laineala: X-lainepikkus, Y-optiline tihedus).
Läbilaskvus on optilise tiheduse pöördväärtus, mida suurem on optiline tihedus, seda väiksem on
pöördväärtus.
Optikakomponendid ei saa olla klaasist kuna see ei ole läbitav IR-kiirgusele, valmistatakse leelis- ja
leelismuldmetallide halogeniidsooladest (NaCl, KBr, CsI). Need neelavad väga vähe infrapunakiirgust.
Vedelate ainete spektri saab määrata kas vahetult või sobivas lahuses. Kuna ükski lahusti ei ole
läbipaistev võivad proovi ja lahusti max-d ja min-d kattuda. Lahustiks on süsiniktetrakloriid CCl4,
kloroform CHCl3,
diklorometaan CH2Cl2; kasutatakse konts lahuseid.
Lahusekiht küvetis on väga õhuke, kuni mm-ni. Kõveti opt.pinnad on valmistatud analoogsest
materjalist nagu optika.
Enimleevinud meetod on uuritava proovi IR-spektri võrdlemine standardaine spektriga. Aine konts
tuleb valida selline et suurima neeldumismax kohal jääks optiline läbitavus vahemikku 5-25%.
Saab uuuritava aine tuvastada farmakopöas, avaldatud IP-spektri abil.
Spektrofotomeeter tuleb kalibreerida, et vältida vigu. Kontrollimiseks kasutatakse 35µm paksuse
polüstüreenkile standardspektrit.
Tänapäeval kasutatakse infrapunaspektrofotomeetrites interferentsi. See on saadud mitte otsese
mõõtmise teel, vaid
Fourier transformatsiooni teel. Tulemus saadakse arvutuslikul teel. Ei lahutada
kiirgust spektriks, vaid läbi proovi suunatakse kogu kiirgus. See kiirgus on peeglite abil
selliselt modifitseeritud , et teatud osa kiirgusest on läbinud erineva teepikkuse. Seetõttu tekib lainete
interferents . Tänapäeval kõik on interferentsmeetodil kasutusel.
1.5. FLUOROMEETRIA.
Põhineb ergastatud molekuli võimel anda osa üleliigset energiat ära valguskvandina. Kui
molekul absorbeerib elektromagneetilist kiirgust ja tõuseb stabiilselt energiatasandilt kõrgemale ebastabiilsele
tasandile, siis ta annab losaenergia ära soojendusenergiana kokkupõrgetel teise molekulidega.
Fluoromeetrias mõõdetakse uuritava aine fluorestsentsi intensiivsust:
Kus F- fluorestsentsi üldine intensiivsus (kvanti/sec) Jo erfastava valhuse intensiivsus (kvanti/sec) C lahuse konts (mooli/l) (E) molaarne neeldumiskoeff b- fluorestseeruva kihi paksus (cm) fluorestsentsi
kvant -saagis (sõltub aine om-st)
Valem kehtib lahuse suhtes, mille opt.tihedus D ei ületa 0,05, millele vastab opt.läbitavus 89%, sest
suure opt.tiheduse juures kiirgus ei levi lahuses ühtlaselt. Lahuse konts peab olema 10-5-10-6, siis
flurestsentsuse ja konts vahel kehtib lineaarne sõltuvus. Suuremate konts puhul sõltuvus kaob ja
ilmneb fluor.nõrgenemine. Lora Sulg, Proviisor II, sügis 2010
Fluor. Intensiivsus sõltub: 1)lahuse omadustest 2)lahuse pH-st, pH mõjutab molekuli konfiguratsiooni 3)lahuses esinevatest kõrvalisest ainetest (O2), võivad väheneda intensiivsust, sest
neelavad osa ergastava valguse energiast või deaktiveerivad ergastanud molekuli. 4)ergastava kiirguse lainepikkusest
Emissioonispekter (tekkinud kiirguse specter) on alati suurema lainepikkusega kui ergastusspekter.
Tavaliselt 50-100 nm võrra pikem. Spektri iseloom on iseloomulikud igale individuaalsele ainele.
Juhul, kui aine ei ole fluorestseeruv, või seda muutuda fluor-
vaks , moodustades derivaadi mõne teise
ainega.
Spektrofluoromeeter.
Tööpõhimõte: valgus elavhõbekvartslambilt (kiirgab valgust teatud lainepikkustel 365nm) või
ksenoonlambilt (kiirgab pidevspektrit) läbib esimese monokromaatori (difraktsioonivõre/valgusfilter)
ning pilu ja langeb küvetile uuritava lahusega. Tekkinud fluor-kiirgus läbib sekundaarse
monokromaatorit ja pilu ning langeb valgustundlikule elemendile, mis registreerub intensiivsuse.
Tavaliselt valgustundlik element on valgusallika ja küveti suhtes 90o nurga all, et vältida erg.kiirguse
sattumist fotoelemendile.
Tulemus esitatakse protsentides.
Enne mõõtmist tuleab apparaat kalibrerida pannakse suurima konts-ga
stand .lahust ja
reguleeritakse mäit nii, et standardi fluorestsentsi intensiivsus moodustaks 90% suhteline skaala
ulatusest.
Kvantitatiivsel analüüsil kasut võrdluslahusena standardaine teadaoleva konts-ga lahust. Paralleelselt
tehakse kontrollkatse, määratakse uuritavat ainet mittesisaldava lahuse foonfluorestsentsi ehk
blanki väärtust. Uuritava lahuse fluorimeetri näidtudest lahutatakse fooni väärtus.
X = [(nproov nblank)*] / [nst nblank]
Kus n - fluorestsents
Sellel meetodil saab tulemust arvutada juhul, kui proovi konts on enamvähem teada ja on võimalik
teha uuritavale proovile lähedase konts-ga stand.lahus (intensiivsused ei tohi erineda rohkem kui 2,5
korda).
Vastasel juhul tuleb mõõta mitme stand.lahuse fluorestsents ja saadud tulemuste alusel arvutada välja
klassikaline kalibratsioonigraafik.
Saab teha ka mitme ainete sisaldava segu fluorestseerumine, isegi juhul, kui kõik ained
fluorestseeruvad. On võimalik valida selline ergastus/emissioonilainepikkuse kombinatsioon või kkonna
tingimused (pH), et fluorestseeruks ainult vajalk aine
1.6 AATOMABSORPTSIOONSPEKTROFOTOMEETRIA.
See on meetod, kus uuritava elemendi aatomid neelavad neile spetsiifilist valguskiirgust.
Antud meetod sobib elementanalüüsiks. Antud juhul ei saa me määrata molekule. Registreerib seda,
kuidas aatomid kiirgust neelavad või kiirgavad.
Kõige primitiivsem aatomspektrofotomeetria on näiteks Na, K määramine, kus mõõtelemendiks on
oma silm. Värvusreaktsioon. Molekulid lagunesid kõrges temperatuuris.
Saades leegist energiat,
läksid aatomid kõrgemale energianivoole ja sealt alla tulles andsid osa energiat ära valguskvandina.
Energiaallikaks soojuskiirgus.
AAS - juhitakse leeki, kuhu on pihustatud uuritav aine, elektromagnetkiirgus. Neeldumisribadel
registreeritakse, kui palju neeldumine vähenes.
Spetsiifilised lambid õõneskatoodlamp.
Plokki on
puuritud auk. Katoodi ees on
anood . Lamp on ise õhutühi.
katioonid kogunevad katoodile (negatiivne ,,-,,),
anioonid kogunevad anoodile (positiivne ,,+").
Kiirgusallikas tuleb valida vastavalt uuritavale elemendile.
Metallide emissioonijooned jäävada rohkem spektri nähtavasse
ossa . Mittemetallidele iseloomulikud
jooned jäävad spektri jäigemasse ossa või isegi alla 180 nm.
Igale elemendile on omane oma komplekt spektrijooni. See on justkui sõrmejälg. Lora Sulg, Proviisor II, sügis 2010
Ajalooliselt vanem on aatomemissioonspektromeetria, kus kõrge temperatuuri mõjul elementide
aatomid lähevad gaasifaasi ja ergastuvad.
Kuna temperatuur mõjutab suhteliselt rohkem ergastatud aatomite hulka, on
leegi temperatuuri
stabiilsus eriti kriitiline antud meetodi korral.
Näiteks Na puhul suureneb leegi temperatuuri muutumisel 10K võrra (2600K -> 2610K) ergastatud
aatomite arv proovis 4% võrra, samal ajal on ergastatud aatomeid vaid 0,02% aatomite koguarvust.
Klassikalisel meetodil kasutatakse proovi atomiseerimiseks
gaasilise kütuse põletamisel
saadavad energiat, enamlevinud on
segud . Õhk-atsetüleen (põlemistemperatuur 2250oC) (õhuhapnik on oksüdeerijaks) või N2O-atsetüleen (2955oC) (kui on vaja kõrgemaid
temperatuure ).
Uuritav aine pihustatakse tavaliselt vesilahusena leeki. Nebuliseerimine ehk udustamine.
Antud juhul leek täidab küveti rolli. Aur läbib leegi. Peale leeki on monokromaator. Valgusallikas ise
kiirgab juba spetsiifilisi spektrijooni. Monokromaatori ülesanne on eraldada proovile langenud
spektrijoontest üks spektrijoon, mis meile huvi pakub. See spektrijoon, millel
soovime mõõtmist
teostada. See langeb fotoelemendile. Pilu laiust on võimalik väga täpselt reguleerida.
Leegis tekib ka teatud kiirgus aatomite ergastumise tõttu, mis summeerub lambi poolt tekitatud
kiirgusega. Aatomite ergastumisest tingitud kiirguse mõju ellimineerimiseks kiirgab lamp valgust
impulssidena, sagedusega 50 impulssi sekundis (50 Hz).
Lambiga on sünkroniseeritud valgustundliku elemendiga seotud võimendi, nii et registreeritakse ainult
lambilt lähtunud kiirgus.
Aatom-spektrofotomeetriat kasutatakse keemiliste elementide (peamiselt metallide) kvalitatiivseks ja
kvantitatiivseks analüüsiks.
On teistest selleks otstarbeks kasutatavatest
meetoditest kiirem ja praktiliselt ei nõua proovi eeltöötlust,
kuna reeglina üks element ei sega teise määramist ning neid on võimali korraga määrata ühes proovis.
I=Io*e-kcl D=log Io/I
I neelava kihi läbinud kiirgus
e - logaritmalus
k kiirguse
neeldumiskoefitsient neeldumisjoone keskel
c neelava komponendi kontsentratsioon
l neelava kihi läbimõõt
Mõõtmist mõjutab ionisatsioon: ioonide kiirgamisvõime ja ioonide võime neelata kiirgust erineb
aatomite omast, keemiliste ühendite teke, mitteselektiivne neeldumine.
Lõpptulemuse saame, nagu spektrofotomeetrias, kasutades kas standardaineid, tehes
kalibratsioonigraafiku (3 standardit, uuritav
proov jääb standardite keskele).
Sobib kitsaks rakenduseks, kuid selles väga spetsiifiline.
3.
KROMATOGRAAFIA Esmalt kujutas Mihhail Tswett, 1903 aastal lahutas komponentideks taimseid värvipigmente, mis
kolonnil oleval sorbendil moodustasid erineva värvusega tsoone.
Krom.lahutamisel jaotuvad segu komponendid kahe faasu vahel, millest üks on liikumatu (suur
eripind -
osakesed sisaldavad väga palju poore või on suure pinnaga) ja teine liikuv faas (
gaas või vedelik mis
filtreerub läbi liikumatu faasi). Lora Sulg, Proviisor II, sügis 2010
Komponentide segu lahutamine põhineb sorbtsioonil gaaside, vedelike või nende lahustunud ainete
seondumine sorbentidele. Sorbentideks on nii
tahked , kui ka vedelad ained.
Absorptsioon seondumine kogu sorbendi mahu ulatuses sorbendiks on vedelik.
Adsorptsioon seondumine sorbendi pinnal sorbeeruvad ained kontsentreeruvad
faaside kokkupuutepinnal. (liikuv vedel faas üuutub kokku liikumatu tahke sorbendiga)
Adrptsiooni
kutsuvad esile molekulaarsed jõud.
Kui sorptsioonil aine ei muutu keemiliselt (säilitab oma individuaalsuse) füüsikaline
sorptsioon .
Protsess on pöörduv, aine või sorbendilt kergesti
vabaneda
desorptsioon .On vähespetsiifiline, võib
kulgeda suvalisel pinnal suvalise aine suhtes. Konst.temp-l intensiivistub gaasi rõhu või aine konts-ni
kasvades.
Kui sorptsioonil toimub sorbendi ja sorbaadi tugev vastasmõju,nii, et nende struktuur muutub
keemiline sorptsioon. Toimub faaside kookupuuutel keemiline
reaktsioon . Desorptsioon on
raskendatud.
Staatiline S. kui S toimub kiiresti ja kvantitatiivselt. Kindel kogus sorbenti segatakse uuritavat
komponenti (komponente) sisaldava lahusega, lastakse toimida kindel aeg ja tahke sorbent eraldatakse
lahusest,
sellelt omakorda eraldatakse uuritav aine (ained) ja neid analüüsitakse edasi. Seda
nimetatakse ka ekstraktsiooniks tahkel faasil. Kasutatkse keerukate segude eelpuhastamiseks enne
põhianalüüsi.
Dünaamiline soprtsioon protsess, mida enamasti kasutatakse kromatograafias. Sorbendile ehk
liikumatule
faasile kantakse uuritava ainete segu. Seejärel lastakse sorbendikihist läbi voolata liikuv
faas. Iga aine sorbeerub vastavalt oma afiinsusele. Mida afiinsem on konkreetne aine liikumatu
faasi suhtes, seda kauem on ta seotud liikumatu faasiga ja seega seda vähem liikuva
faasiga.(peitub sorbendikihis). Peetunud molekulid ei l'he liikuva faasiga kaasa ega ei osale liikumises
seni, kuni oile desorbeerunud tahkelt faasilt. Mida rohkem on aine seotud liikumatu faasiga, seda
aeglasemalt see liigub. Mida kauem aine viibib liikuvas faasis, seda kiiremini läbib ta liikumatu faasi,
ainete liikumiskiiruse
kolonnis määrab otseselt ära liikuvas faasis viibitud aeg.
Kromatografeerimise tingimuseks on teatav afiinsuse tasakaal liikumatu ja liikuva faasi vahel.
Sorbeerumise intensiivsus sõltub kolmest asjast:
polaarsus . Liikumatule faasile sorbeerumine oleneb
aine, liikumatu faasi ja liikuva faasi polaarsusest. Mida erinevam on aine polaarsus liikumatu faasi
polaarsusest, seda kiiremini läbib ta kolonni e. seda väiksem on afiinsus ja seda rohkem aega viibib
liikuvas faasis. Mida sarnasem on liikuva faasi polaarsus liikumatu faasi polaarsusele, seda suurem
elueeriv jõud ehk võime kanda ainet läbi liikumatu faasi. See tähendab, et kromatografeeritav aine ja
liikuv faas konkureerivad üksteisega liikumatu faasi pinna pärast. Mida lähedasemad on liikumatu faasi
ja uuritava aine polaarsused, seda paremini
seondub aine liikumatule faasile.
3.1 Kromatograafia liigid.
Agregaatoleku järgi
1)
gaaskromatograafia liikuvaks faasiks on sobiv gaas, nt lämmastik,
vesinik või heelium.
Solvendiks võib olla vesi, benseen vms;
2)
vedelikkromatograafia ;
3) gaas-vedelikkromatograafia tegemist kromatograafia eriliigiga.
Kolonn on täidetud tahke
kandja osakestega, millele on
kantud vedel faas. Absorbeerub vedelas faasis, mitte tahkel.
Liikumatu faasi ja aine vastasmõju järgi
1) molekulaarkromatograafia see on kromatograafia liik, millest enamasti räägime. Sorbaat on
sorbendile sorbeerunud aine ehk aine, mida soovime lahutada. Sorbendi ja sorbaadi vaheline mõju on
üsna nõrk ehk tegemist on füüsikalise sorptsiooniga. Siia alla kuuluvad: adsorptsioonkromatograafia see on klassikaline kromatograafia. jaotuskromatograafia see
mehhanism toimub kapillaarkolonnis gaaskromatograafia
korral. Pikk kvartskapillaar 20-30 m pikk läbimõõduga 50-150 m. kapillaari sein on kaetud
vedelikukihiga. Valendikus toimub gaasi liikumine. Aine absorbeerub vedelikkukihti.
Jaotumine vedelfaasi ja gaasifaasi vahel; Lora Sulg, Proviisor II, sügis 2010
molekulaareksklusioonkromatograafia (
geelkromatograafia ) size-exclusion
chromatography. Ehk suuruse järgi ellimineeriv kromatograafia. Väiksemad osakesed, mis läbivad
kapillaare, läbivad pikema tee. Suuremad osakesed jõuavad kolonnist enne välja.
2) Kemosorptsioonkromatograafia tekivad keemilised reaktsioonid aine, sorbendi ja eluendi
vahel, mis võivad olla pöördumatud või pöörduvad.
ioonvahetuskromatograafia ioonvahetuskolonniga võimalik ioniseeruvaid aineid lahutada.
Kolonni liikumatu faasi pind sisaldab nt. katioone. Need tinglikult hoiavad kinni
anioone . Mida paremini
hoitakse anioone kinni, seda aeglasemalt
anioon liigub. afiinsuskromatograafia seda kasutatakse enamasti
bioloogiliste objektide korral.
Antikehad seotakse liikumatule faasile. Kolonnist lastakse
veri läbi.
Antikeha seondub vastava
molekuliga ja see seotakse kolonni. Järgmises faasis elueeritakse sobiva solvendiga. Eelis on see, et
on väga tundlik ja spetsiifiline. Võib olla 500 erinevat molekuli, aga ainult 1 molekuli suhtes afiinne ning
seda on võimalik eraldada. Aga seda meetodit kasutatakse vähe, vaid teatud bioloogiliste
proovide korral.
Liikumatu faasi paigutuse viisi ja voolutustee ristlõike geomeetrlise vormi järgi
1) kolonnkromatograafia klassikaline vedelikkromatograafia kolonnkromatograafia.
Vedel faas liigub läbi kolonni rõhkude vahe tõttu kolonni sisendil ja väljundil. Ühemeetrine
kolonn, rõhkude vahe sisendil ja väljundil 0,1 atm. 10 m 1 atm. Võimalik kasutada ka
pumpa kõrgsurvevedelikkromatograafia korral. Rõhk sadades atm-s. kolonn on materjalist, mis suudab rõhule
vastu panna, tavaliselt
terasest .
2) kapillaarkromatograafia täidise osakeste läbimõõt on samas suurusjärgus kolonni
läbimõõduga. Kapillaari läbimõõt plaadile . Vedelik (liikuv faas) liigub mööda liikumatu faasi ristlõikepindala. Tegemist kahemõõtmelise
plaadiga. Soovitatakse kasutada plaati, kuid on ka paber, nt. tavaline filterpaber. Tselluloosi kiud
moodustavad liikumatu faasi ja meid huvitavad ained seostuvad tselluloosiga pikkade
tärklisemolekulidega. Sõltuvalt osakese
suurusest kihtkromatograafiat jaotatakse: tavaline osakeste läbimõõt 200-250 m; kõrgefektiivne osakeste läbimõõt 1-25 m. Suhteliselt laialt kasutusel, sest on odav ja lihtne kasutada. Vaja ainult plaati,
hermeetiliselt suletavat klaasanumat. Kuid pole niivõrd informatiivne kui teised kromatograafid. Liikuva faasi liikumine toimub vastupidiselt raskusjõule. Paberiga tavaliselt
alanev meetod.
Kromatograafia eesmärgi järgi
1) analüütiline kromatograafia eesmärk kas
kvalitatiivselt määrata teatud ainete olemasolu
segus või kvantitatiivselt määrata ainete kogus. Aine otseselt ei huvita. Ainult tema identsus või
sisaldus.
2) preparatiivne kromatograafia eesmärk on eraldada segust teatud aine. Võidakse kasutada
ka 2-3 m läbimõõduga kolonne.
AINE SEGU KOMPONENTIDE KVANTITATIIVNE SISALDUSE SPPEKTROFOTOM.MÄÄRAMINE
(UV ALAS)
Vastavalt Lambert-
Beeri seadusele sumeeruvad kõigi segu komponentide optilisd tihedused antud
lainepikkusel ja seega mõõdetakse summaarset optiliset ithedust. Selleks, et määrata individuaalsete
komponentide kvantit sisaldust:
1)peavad komponentide neeldumimax-d piisavalt erinema üksteisest. Mida suurem on erinevus,
seda täpsem tulemus saadakse.
Soovitav oleks, et ühe komponendi neeldmax
langeks kokku teise
komponendi neeldmin-ga. Ideaaljuhul ühe kompon neeld.maksimumis teise komponendi opt.tihedus 0.
Siis saaks
loobuda teise komponentide mõju arvestamist ja analüüsida segu nagu see sisaldaks vaid
määratavat ainet
2)komponentide lahuste konts-d peavad olema päris väikesed, et kõikide nende korral optiline
tihedus oleks lineaarses sõltuvuses kontsentratsiooniga. On vaja registreerida erineeldumisnäitajaid Lora Sulg, Proviisor II, sügis 2010
erinevatel
kont -del lineaarse sõltuvuse juhul ei sõltu erineelnäit konts-st ja
graafikud langevad kokku.
Komponendi konts-n arvutatkse selle erineeldnäitajast.
Ühekomponentse segu korral leitakse konts-ni selle erineeldnäit ja antud lainepikkusel:
Kus a erineeldnäitaja; b lahusekihi paksus, D optiline tihedus
PLANAARKROMATOGRAAFIA.
On kromatograafiline protsess, mis kulgeb liikuva faasi liikumisel inertsele pinnale kantud sorbendi
õhukeses ihis.
Liikumatuks faasiks on thake sorbent (silikageel). Mehhanism põhineb adsorptsioonil.
Liikuv faas kandub sorbendikihis edasi tavaliselt kapillaarjõudude mõjul.
Moodustavad ained kandjal ümmargused või ovaalsed laigud. Laikude kogumit nimetatakse
kromatogrammiks.
Retensioonifaktor Rf iseloomustab aine liikuvust kromatografeerimisel. Kujutab endast uuritava aine
ja liikuva faasi keskmiste liikumiskiiruste suhet kromatogrammi saamise aja jooksul. Rf väärtusele
mõjutavad tugevasti kromatografeerimistingimused. Kasutatakse orienteeruvaks samastamiseks.
Rf = a/b , kus a-kaugus stardijoonest laigu keskpunktini; b kaugus stardijoonest liikuva faasi
frondijooneni.
Rs vähetundlik juhuslikele kõrvalekallete suhtes, kasutatakse täpsemale kromatografeerimise kiiruse
hindamiseks. (standardi suhtes). Kujutab ennast ühe aine Rf väärtuse suhet standardiks võrtud aine Rf
väärtusesse. Valitakse standard nii, et Rs jääks piiridesse 0,5-2. Kasutatakse orienteeruvaks
samastamiseks
Rs = Rfproov/Rfstandard..
Tegelik samastamine toimub analüüsitava aine ja sama aine autenstse näidise üheaegsel kromatograf-l
ühel plaadil. Kui
proovid on identsed, on laikude valiums ühesugune ja neil on võrdsed Rf väärtused.
Segu kui tegu on sama ainega, siis peab kromatogrammil ilmuma vaid üks laik.
Kromat-seks tuleb valida selliseid tingimused (sorbendi materjal ja eluendi koostis) et Rf väärtus
erineks nullist (aine ei lahku stardijoonelt) ja ühest (aine liigub frondijooneni). Lisanditel ja põhiainel
peavad olema erinevad Rf väärtused.
KROMATOGRAMMI ANALÜÜS.
Kromatogramm kõver, mis moodustab rea piike nii palju pike, kui palju oli uuritavaid aineid.
Esimene
piik on liikumatu faasiga mitteseondunud aine piik.
Retensiooniaeg (tR) aeg, mis kuulub proovi viimisest kolonni kuni piigi
maximum kromatogrammil
ilmumiseni. Sõltub liikuva faasi liikumiskiirusest.
Surnud aeg (t0) aeg, mil proov oli kolonnis lahustutuna liikuvas faasis kuid pole sorbeerunud
liikumatule faasile.
Korrigeeritud aeg (t`R = tR t0)
Retensiooniruumala (VR = tRu) võrdub retensiooniaeja ja liikuva faasi mahtkiiruse u korrutisega. korrigeeritud ruumala.
Suhteline retensioon (tsuht ) korrigeeritud analõõsitava komponendi ja standardina kasutatava aine
retensiooniaegade suhe. Suurus sõltub liikumatu ja liikuva faasi omadustest ja temp-st, ei sõltu liikuva
faasi mahtkiirusest, sorbendi hulgast ja kolonnipikkusest. Seetõttu, suurused tsuht, t´R ja v´R
reprodutseeritavad ja võimaldavad identifitseerida uuritavat ainet. Lora Sulg, Proviisor II, sügis 2010
Retensioonkoefitsient (R) uuritava aine kiiruse suhe liikuva faasi kiirusesse. Näitab, milline osa
retensiooniajast asub uuritav aine liikuvas faasis. T0 ja tS on aine viibimise ajad liikuvas ja liikumatus
faasis.
Jaotuskoefitsient (D = Cs/C0) kromatografeeriva aine jaotumist liikumatu ja liikuva faasi vahel. CS ja
C0 on aine konts võrdsetes hulkades vastavalt liikumtus ja liikuvas faasis.
Mahtuvuskoefitsient (k´) näitab, kui tugevasti uuritav aine seondub sorbendiga ja kiu kaua ta sellel
viibib võrreldes liikuva faasiga. Optimaalseks väärtuseks on 1.5-4.
Lahutuvuskriteerium iseloomustab piikide lahutuvust. Piikide kattumine on lubatav vaid teatud
piirini, vastasel juhul ei saa täpselt arvutada nende parameetreid ja seetõttu ka aine sisaldust proovis.
Kus w2 ja w1 on kahele ainele vastavate piikide
laiused baasijoonel.
Kui piikide laiused on võrdsed (w2=w1=w):
Euroopa farmakopöa järgi:
Kus tR1 ja tR2 retensiooniajad või teepikkused piki baasijoont alates süstehetkest kuni kahe
kõrvutioleva piigi max-dest tõmmatud vertikaaljoonteni; wh1 ja wh2 piikide laiused poolel kõrgusel.
Teoreetiliste taldrikute teooria põhineb
eeldusel , et kromatografeeritav aine läbib sorbendkihi
portjonite kaupa ja seejures jaotub võrdeliselt selle elementaarlõikudel taldrikutel. Iga uus liikuva faasi
portsjon , mis liigub esimesele taldrikutele, kutsub esile aine uue jaotumine liikuva ja liikumatu faasi
vahel ja osa selle ülekande teisele taldrikutele. Nüüd saabub teasel
taldrikul aine taasakaalujaotus
tahke ja vedela faasi vahel ja uus liikuva faasi portsjon kannab osa ainet kolmandale taldrikutele.
Mida kauem see toimub, seda rohkem arvu
taldrikutega on aine seotud.
Teor taldtikute arvu N võib kasutada selle hindamiseks kui palju kromatografeeritavale ainele vastav
tsoon sorbendikihil laiali valgub ehk kolonni effektiivsust ehk lahutusvõime hindamiseks. Mida vähem
uuritav aine sorbendikihil hajub, seda suurem on taodrikute arv ja kolonni lahutuvusvõime.
Retensiooniaeg on proportsionaalne teor taldrikute arvuga N . t´R = kN, kus k proportsionaalkoefitsient.
Kus w piigi laius 1/20 kõrgusest.
Euroopa farmakopöa järgi:
Sümmeetriafaktor (As) :
Kus w0,05 piigi laius 1/20 kõrgusest; d vahemaa piigi maksimumkõrgusest tõmmatud vertikaaljoone
ja piigi esiserva vahel. Väärtus 1,0 tähistab täielikku sümmeetriat.
Signaali-müra suhe tähistab piiri tundlikkust ja mõjutab kvantifitseerimise täpsust: Lora Sulg, Proviisor II, sügis 2010
Kus H uuritavale ainele vastav piigi kõrgus standardaine kromatogrammil, mõõdetuna piigi
maksimumist kuni ekstrapoleeritud baasijooneni, mis on leitud 20 kordsele laiusele poolelt kõrguselt
vastavalt baasijoone lõigust arvutatud keskmisest signaalist
h- taustamüra
amplituud kromatogrammil saadud peale blangi süstimist, arvutatuna teepikkuselt, mis
võrdub 5-kordse piigi laiusega poolkõrguselt vastava võrdlusaine kromatogrammilt, võimaluse korral
piihi asukohale vastavast
piirkonnast .
Selleks, et identifitseerida ainet, peab segnaali-müra suhe olema vähemalt 3, selleks, et määrata
kvantitatiivset sisaldust, peab signaali-müra olema väh 10.
Piigi pindala arvutamine.
1.piigi moodustava kõvera käänupunktidest joonistatkse puutujaid, mis moodustavad kolmnurga.
Pindala leitakse kolmnurga valemist: A=wh/2, kus w piigi laiu baasijoonel, h piigi kõrgus. Meetodi
viga on 3%.
2. leitakse korrutades piigi
laiuse poolel kõrgusel wh piigi kõrgusega h: A=whh. viga on 6%. Lihtsaim ja
enimkasutatavaim.
3. Condal-Boshi meetod. Liidetakse piigi laius w15 15% kõrguselt laiusega w85 85% kõrguselt,
jagatakse kahega ja korrutatakse piigi kõrgusega h. Viga on 0,5%, on sobivam ebasümmeetrilise piigi
pindala leidmiseks.

























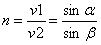
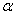
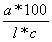
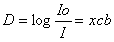
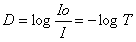
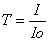
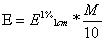
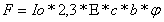




Kõik kommentaarid