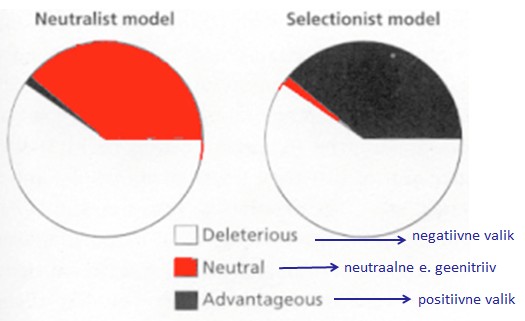
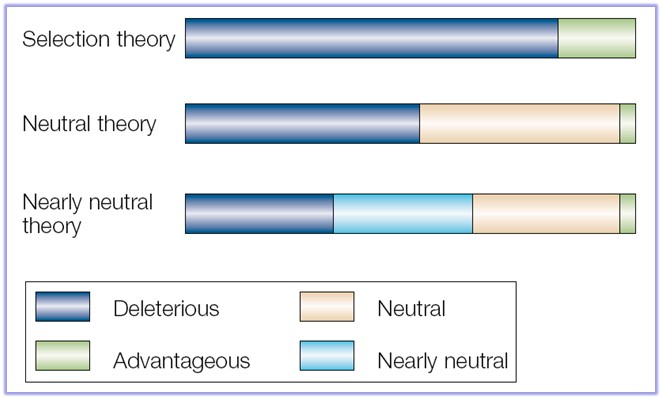
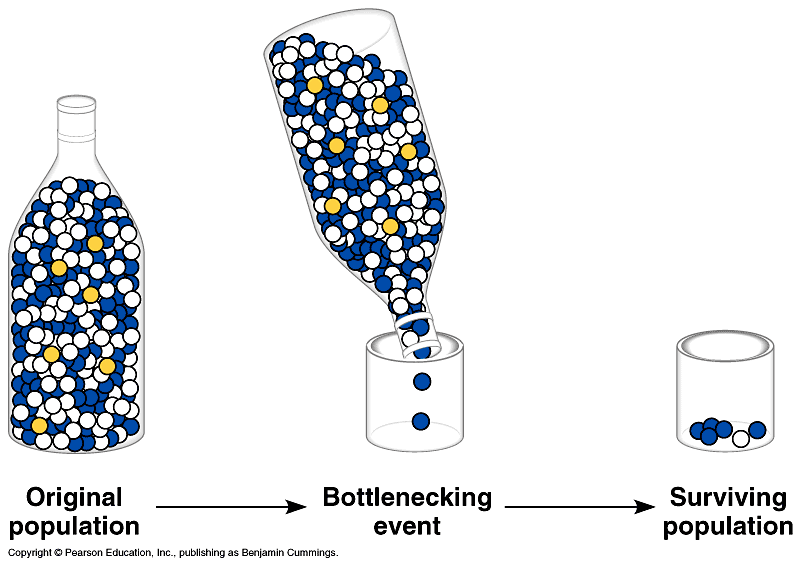
Väiksem sagedus suurendab kadumamineku ja suurem sagedus fikseerumise tõenäosust. 5. Kuidas mõjutab (kasvatab või kahandab) juhuslik geenitriiv geneetilist diversiteeti populatsioonide sees ja vahel? Geenitriiv kahandab geneetilist diversiteeti populatsioonisiseselt ning kasvatab mitmekesisust populatsioonide vahel. Nt väike isoleeritud populatsioon võib lahkneda läbi geenitriivi mõju suuremast populatsioonist. 6. Seleta mõistet molekulaarne kell. Kui konstantne üldiselt selle kiirus on eri organismide vahel ja millest see sõltub? Molekulaarne kell aitab määrata evolutsiooni ajalist kulgu DNA nukleotiidide ja aminohapete järjestuste põhjal. Igal liigil tekivad mutatsioonid erineva sagedusega ja seda mutatsioonide tekkesagedust nimetataksegi molekulaarseks kellaks. Molekulaarne kell on konstantne ehk mutatsioonid fikseeruvad konstantse kiirusega. Tegelikkuses eri liikide vahel päris
sees ja vahel? [Katre] Mida väiksem geneetiline diversiteet (st väga erinevad alleelisagedused), seda väiksem on triivi mõju. Triiv aga on juhuslikud muutused alleelisagedustes põlvkonnast põlvkonda, mis on tingitud populatsiooni suuruse lõplikusest. Geneetilise triivi tagajärjel võib osa alleele (geenivariante) kaduda, niiet geneetiline mitmekesisus väheneb. Geenitriivi mõju on seda suurem, mida väiksem on populatsioon. 6. Seleta mõistet molekulaarne kell. Kui konstantne üldiselt selle kiirus on eri organismide vahel ja millest see sõltub? Molekulaarne kell on molekulaarse evolutsiooni konstantsus. Tegelikkuses eri liikide vahel päris konstantne ei pruugi olla. Sõltub näiteks paljunemisviisist (suguline või mitte-suguline). ME. Kell ei ole piisavalt konstantne - piisab kui populatsioonisuurused ajas muutuvad, et kell ka kaasa jõnksuks. Sama loogika: kui populatsiooni suurus langeb muutub üha suurem osa kergelt kahjulikke mutatsioone
Statistilised meetodid, et hinnata LV mõju pidevatele tunnustele populatsioonis. Saltatsionistid eitasid LV-d ja arvasid, et muutused pidid toimuma hüppeliselt, kuna vahepealsed vormid ei olnud nende meelest kohased ja LV peaks neid siis elimineerima. Lamarkism oli valdav omandatud tunnused on päritavad, liigid muutuvad, kuid ei jagune ega kao. Primitiivseid liike tekib juurde. Sisemine täiustumistung, sunnib saama järglasi, kes on temast paremad. Darwinil oli kaks seotud teooriat evolutsioon (liikide muutumine ja jagunemine, paljude liikide väljasuremisega) ja LV (kui evolutsiooni mehhanism). Evolutsioon on hargnemine vastavalt kohanemine uute keskkonnatingimustega. Puudub vajadus hierarhiale, lihtsalt keerukale. 9. Miks ei leidnud Darwini idee LV-st tema kaasajal toetajaid? Usuti, et liigid muutuvad, aga ei usutud, et need toimuvad sammhaaval. Kuna erinevused eri kehaosade vahel on liiga erinevad, vahepealsed vormid ei saa olla kohased. Kuna pärilikkust ei
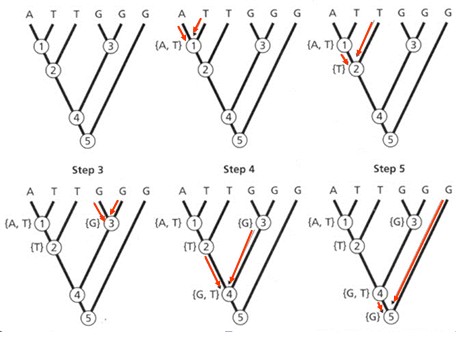
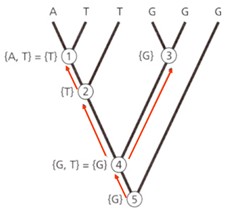
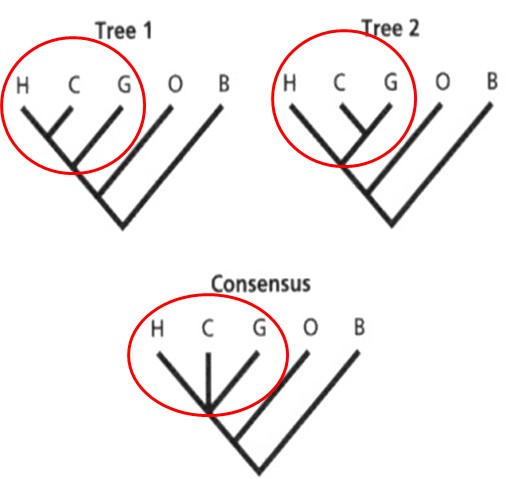
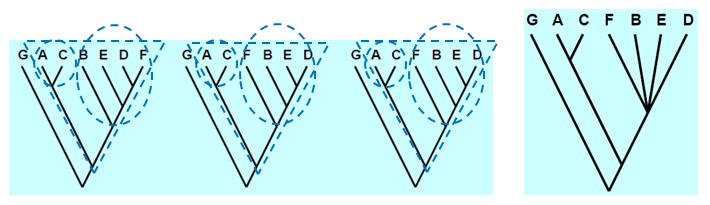
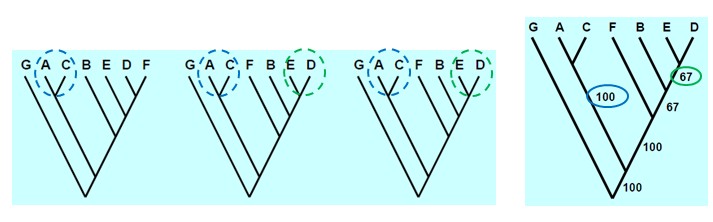
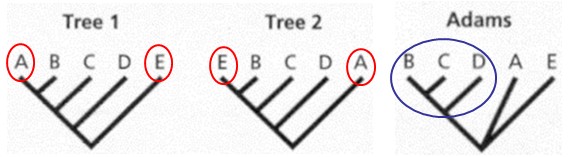
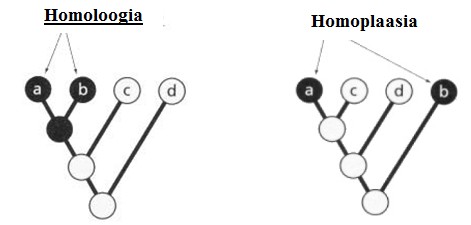
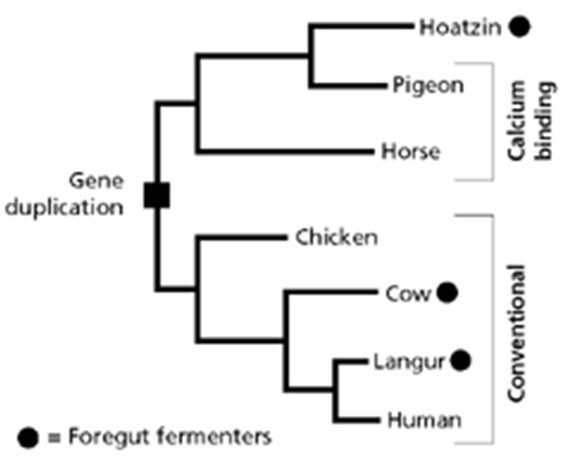
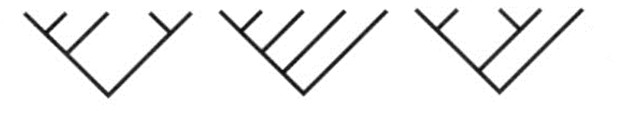
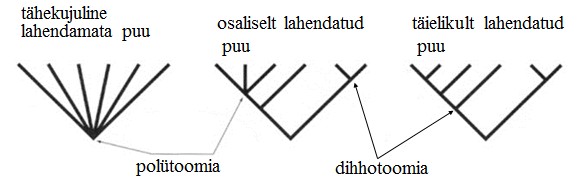
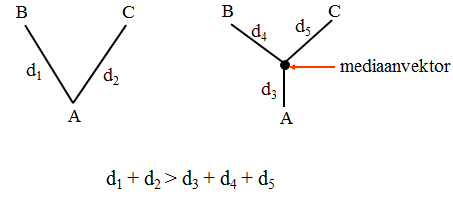
seoses teise funktsiooni kaudu realiseerunud kohasusega Nt taimedel vegetatiivne ja generatiivne paljunemine piiratud sama ressursi poolt. 27. Hõimuvalik is the evolutionary strategy that favours the reproductive success of an organism's relatives, even at a cost to the organism's own survival and reproduction. 28. Koevolutsioon on kahe või enama vastassuhteis oleva liigi (või muu bioloogilise üksuse) üksteisest sõltuv evolutsioon. 29. Kladistika ehk fülogeneetiline süstemaatika on fülogeneetilise süstemaatika peamisi meetodeid. See on süstemaatika üks põhisuundi, mis baseerub taksonite fülogeneesiarvestamises.Ei tugine üksnes liikidevahelise sarnasuse mõõtmisele, vaid võtab aluseks liikide eristumise. Kladistikas koostatakse fülogeneesipuid, kus on esitatud evolutsiooniline "elu puu". Kladistika vaike-eeldusteks on
Ehk keskkonnatingitus. Kvantitatiivse tunnuse muutlikkuse osa, mis on tingitud keskkonna mõjudest organismidele populatsioonis. 108. Valiku meetodid (individuaalne ja massvalik). Individuaalne valik Nt ühe teatud punapea välja valimine. Fenotüüpide ennustamine vanemate keskväärtuse alusel. Massvalik Nt kõikide punapeade välja valimine jne. Selektsiooni diferentsiaal (S), selektsioonikoefitsient (s), valiku intensiivsus, selektsiooni tulem (R). 109. Molekulaarne mimikri. Insuliinist sõltuv diabeet, insuliinist sõltumatu diabeet. 110. Sünteensus. Inimesel ja hiirel üle 200 homoloogse DNA piirkonna. 111. Sisearetus. Ehk inbriiding. Selliste isendite ristamine, kelle geneetiline sugulus on suurem kui populatsioonis keskmiselt. Sisearetus on nt sugulasabielud, Amishi religioosne pere, Euroopa ''siniverelised'' (kuningaperekonnad). 112. Välisaretus. Ehk autbriiding. Taime- ja loomaliikide, kes
Sellega sai evolutsiooniteooria aluseks populatsioonigeneetika. Neodarvinismi kohaselt on Darwini poolt võimalikuks peetud pangeneetiline pärandumine (s.t., et ka elu jooksul omandatud tunnused võiksid päranduda järglastele) võimatu. Geneetiline tekib muutlikkus populatsioonides juhuslikult mutatsioonide tõttu ja rekombinatsiooni tõttu (see on homoloogiliste kromosoomide ristsiire meioosi ajal). Evolutsioon seisneb põhiliselt alleelisageduste muutumises põlvkondade lõikes geenitriivi, geenisiirde ja loodusliku valiku tagajärjel. Liigiteke leiab aset järk-järgult, kui populatsioonid geograafiliste barjääride tõttu reproduktiivselt isoleeruvad. Postulaadid: Populatsioonid sisaldavad geneetilisi variante, mis tekivad juhuslikult mutatsioonide ja rekombineerumise tulemusel (st. mitte adapteerumisest tingituna);
seerimise teooria ja praktika. Põhja-Ameerika bioloogidel on enamasti vas- tupidi: taksonoomia hooleks on teooria, süstemaatika alla käib praktiline klassifitseerimistöö. Nähtavasti on õigem loobuda nendele kahele terminile erineva tähendusvarjundi andmisest. (Märkigem, et paljud bioloogia üldised terminid on küllalt noored. Isegi termin bioloogia on kasutusel alles alates 1800. aastast - Burdach, La- marck; ökoloogia termini tõi kasutusele 1866. a. Haeckel. Evolutsioon tuli kasutusele mitte Darwini, vaid Spenceri töödega.) Süstemaatika ehk taksonoomia alla käib organismide klassifitseerimine, identifitseerimine ja nomenklatuur (nimetuste andmine). Nende ülesannete täitmiseks on vajalik ka evolutsiooniprotsessi uurimine (varieeruvus, lii- giteke, hübriidide moodustumine jm.) ning fülogeneesi (viisid, aeg, koht) selgitamine. Süstemaatika rakendusaladeks on identifitseerimiseks ja klassifitseeri-
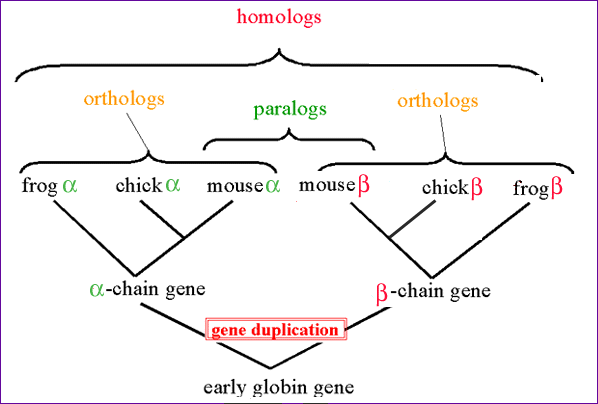
kuid nad on erinevas keskkonnas, nende puhul väljendab korelatsioonikeoefitsent laiatähenduslikku päritavust, kui suur mõju on keskkonnal tunnuse avaldumises 348. Valiku meetodid (individaalne ja massvalik): individuaalvalik- järglastest valitakse ristamiseks välja konkreetsed isendid, kasutades fenotüüpide ettearvamise meetodeid, massvalik- ristamiseks valitakse vanemate hulgast välja kindlate omadustega väike grupp isendeid 349. Molekulaarne mimikri: bakterite ja viiruste pinnamolekulid (antigeensed) võivad sarnaneda peremeesorganismi rakkude membraanimolekulidega, mida immuunsüsteem ei ründa, nt antikehad, mis toodetakse veise albumiini vastu, hävitavad ka B- rakke 350. Sünteensus: 1. Klassikalises geneetikas tähendab geenide ja muude geeniliste elementide paiknemist samas kromosoomis, kuid vaatamata füüsilisele aheldatusele ei pea nad olema suure vahemaa tõttu geneetiliselt aheldatud 2. Genoomikas tähendab










































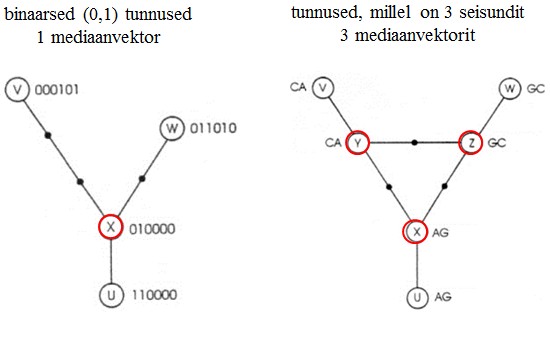
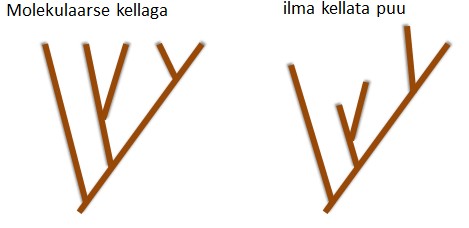
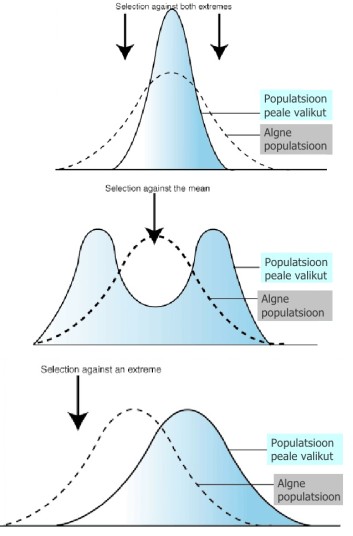
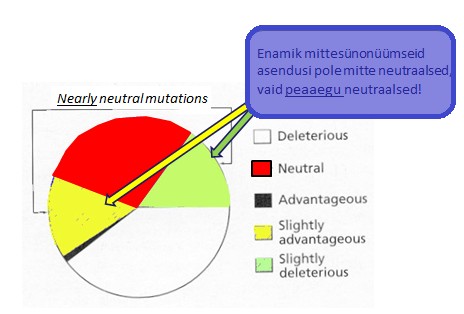

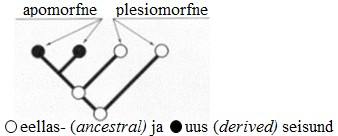
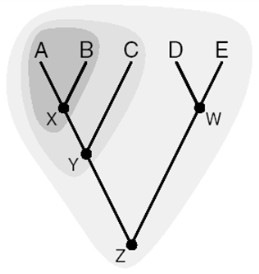
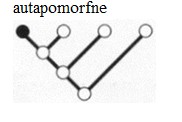
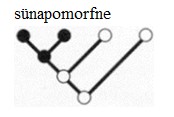
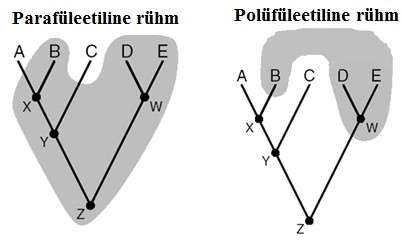
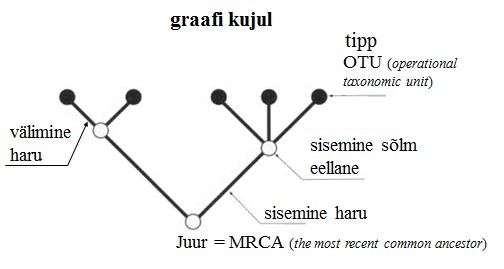
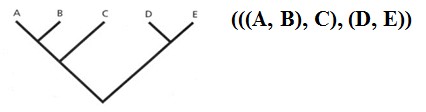
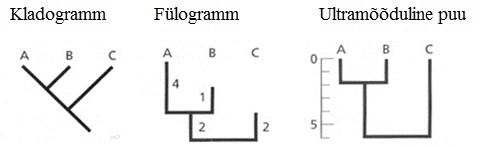




Kõik kommentaarid