TARTU
ÜLIKOOL
LOODUS-
JA TEHNOLOOGIATEADUSKOND
MOLEKULAAR -
JA RAKUBIOLOOGIA INSTITUUT
Koostu tegemine algusest peale: mida iga bioloog peaks teadmaReferaat
TARTU
2014
SissejuhatusEsimene
täielikult sekveneeritud
genoom kuulub bakteriofaagile Φ-X174. Edukas viirusegenoomi DNA järjestuse kindlaks tegemine kannustas
ette võtma mahukamaid projekte ja üks kulukamaid nende seas on
„Inimese genoomi projekt“ (HGP, inglise keeles
the
Human Genome Project ).
Tänaseks on välja töötatud järgmise põlvkonna
sekveneerimismeetodid (NGS, inglise keeles
next-generation
high-throughput sequencing),
mis nõuavad üha vähem
ajalisi ja rahalisi ressursse. Selline
soodus olukord on vallandanud sekveneerimisbuumi, mis on tõstatanud
uue probleemi – mida teha saadud
andmetega ? Üha rohkem tuntakse
vajadust sekveneeritud DNA järjestusi analüüsida ja tõlgendada. Paralleelselt sekveneerimismeetodite arenguga proovitakse välja
töötada üha täiustatumaid
programme andmete töötlemiseks.
Spetsiifilisele andmemassiivile sobiva programmi leidmine on oluline
ülesanne võimalikult täpseks andmetõlgenduseks.
Keeruliseks
on osutunud ülesanne leida selline kombinatsioon
sekveneerimisandmetest ja arvutialgoritmidest, mille väljundiks
oleks võimalikult kõrge kvaliteediga koostu (
assembly ).
Koostu peab olema kõrgkvaliteediline, sest vastasel juhul osutuksid
selle informatsiooni põhjal tehtavad järjeldused vääraks. Genoomi
funktsionaalsete elementide
ennustamine , kodeerivate geenide
ülesleidmine, evolutsioonilise päritolu
selgitamine – kõiki neid
protseduure mõjutab oluliselt koostu kvaliteet.
Koostu
kvaliteedi probleemi muudab komplekssemaks järjest uuemate
sekveneerimistehnoloogiate väljatöötamine. Uued sisendandmed, mis
söödetakse programmidele, on teistsuguste iseärasuste ja
vigadeprofiiliga kui sekveneerimise teerajajaks osutunud Sangeri
meetodi
lugemid (
read).
Seega peavad tarkvarade
loojad pidevalt olema arengutega kursis ja
kohastuma nii, et loodavad programmid suudaksid andmeid õigesti
töödelda. Kuna sisendandmed on kiires muutumises, on keeruliseks
osutunud ka
seniste koostute kvaliteedi hindamine. Kui genoom saab
assembleeritud ehk taas kokkupandud, mille alusel võivad teadlased
väita, et tegemist ongi absoluutselt õige koostuga? Meetrikud,
mille alusel antakse koostutele kvaliteedihinnanguid ei ole
sugugi mitte alati üksteist võimendavad. Esineb lõivsuhe õigsuse (
vigade puudumise) ja pikkuse parameetrite vahel. Järgmiseks oluliseks
hetkeks on otsuse langetamine,
kumb iseloomustaja on olulisem, kas
pikkus või vigade puudumine?
Koostu
tegemine algusest peale ehk de
novo assembly
Koostuid
on võimalik üles ehitada kahel
erineval viisil. Vastavalt sellele,
kas on olemas referents-
sekventsid ,
jagatakse koostuid referentsi alusel koostatuks või
de
novo-ks
(ld „uus“).
De
novo
lähenemisviisi juures on tegemist organismiga, kelle enda ja
lähisugulaste genoomi ei ole veel sekveneeritud. Teine lähenemisviis
kasutab võrdlust, mille puhul kasutatakse lähisugulase
sekveneeritud genoomi koostu tegemise protsessi giidina. Siiski kogu
genoomi jaoks seda kasutada ei saa ja unikaalsete järjestuste jaoks
tuleb rakendada
de
novo
lähenemisviisi.
De
novo
genoomi koostu koostamine on ajaliselt ja rahaliselt kulukam, sest
eelduseks on suur
mate- pair raamatukogu
olemasolu .
Esmalt on vaja lähteandmeid, mida pakub DNA
sekveneerimine . Sekveneerimine
(inglise keeles
sequencing)
tähendab bioloogilises ja biokeemilises kontekstis meetodit, mis
võimaldab määrata biopolümeeride (valkude ja nukleiinhapete)
primaarstruktuuri. Sekveneerimise tulemiks on sekvents, mis sisaldab
endas kindlatest märkidest
koosnevat biopolümeeri jada. Tänasel
päeval on sekveneerimine jagunenud kahte suunda - esimene (Sangeri
ahela terminatsioon) ja järgmine põlvkond (NGS, inglise keeles
the
next generation sequencing).
Nõudlus efektiivsemate tehnoloogiate järele tekkis juba HPG kestel.
Sangeri meetod on kahtlemata oluline saavutus bioteaduste vallas,
kuid perspektiivitu - meetod nõuab mahukaid ajalisi ja rahalisi
ressursse. Nüüdseks on arendatud Sangeri meetodist järgmise
põlvkonna sekveneerimismeetodid. Sünonüüm NGS-le ehk
massiivne paralleelne sekveneerimine tähendab antud meetodi puhul, et terve
genoom eraldatakse väiksemateks ühikuteks ning seejärel
ligeeritakse adapterjärjestusele DNA sünteesi käigus –
sekveneerimine ja DNA süntees toimuvad üheaegselt.
Sekveneerimisprotsess on muudetud kiiremaks, odavamaks ja täpsemaks.
Pärast
genoomi sekveneerimist on järgmiseks etapiks koostute (
assembly)
ülesehitamine.
Kui genoomi sekveneerimine andis informatsiooni
nukleotiidse järjestuste kohta, siis koostute arvutiprogrammid
proovivad rekonstrueerida erinevaid pikemaid biopolümeeride
järjestusi kasutades selleks sekveneeritud järjestuste joondamist
mitmesuguste algoritmide alusel. Assemblerid tööpõhimõte eeldab, et kui kahel lugemil esineb
nukleotiidiline
ühisosa , siis pärinevad nad tõenäoliselt genoomi
samast kromosoomi
piirkonnast . Kui selline kattuvus on tuvastatud,
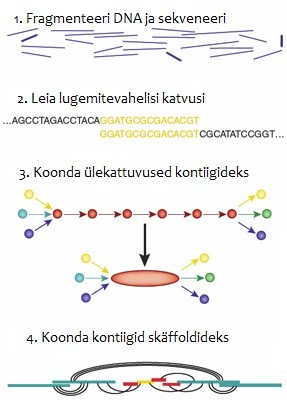
siis joondab programm lugemid vastavalt kontiigideks. Kontiig
ehk piirnevaid järjestusi moodustavad komplekt ülekattuvaid DNA
segmente (lugemid), mis üheskoos moodustavad ühe DNA regiooni
konsensuspiirkonna.
Kontiigide
puhul ei ole teada, kummast DNA
ahelast on nad rekonstrueeritud.
Asukoha määratlemiseks viiakse kontiigidega läbi protseduur, mida
nimetatakse skäffoldimiseks. Skäffoldimisetapil kasutatakse kahte
tüüpi järjestusi: kontiige (liidetud lugemeid) ja
lugemite paare (
mate
pair). Toimub
kontiigide järjestamine ning orienteerimine kasutades lugemite
paarist pärit lisainformatsiooni, mis leidub nende järjestuse
mõlemas otsas (
paired end). Paariliste
lugemite tegemisel fragmenteeritakse DNA ja saadakse järjestusi,
mille otsi sekveneeritakse – genereeritakse lugemid. Fragmentide
keskmine piirkond jääb tavaliselt sekveneerimata. Skäffold
koosneb kontiigidest ja kontiigide vahele jäävatest tühimikest
(
gaps).
Paralleelselt termini
paired
end kõrval
kasutatakse ka
mate
pair, mis täidavad
mõlemad sama eesmärki ehk annavad informatsiooni kahe lugemi
vahelisest füüsilisest kaugusest. Erinevus nende vahel seisneb
raamatukogu tegemise metodoloogias. Kuigi tühimiku (
gap)
järjestus ei ole teada, on võimalik hinnata selle pikkust kontiigide endi pikkuste järgi, mis on
umbkaudu teada.
Koostuprogrammid
jagunevad skäffoldimisetapi läbiviimisel kahte erinevasse suunda.
Esinevad nii kaks-ühes koostud, mis sisaldavad juba
skäffoldimismoodulit, kuid nende kõrval esineb programme, mis on
just spetsiaalselt skäffoldimise jaoks disanitud. Kuigi pakub
kaks-ühes programm kasutajamugavust, ei ole see alati kõige õigem
viis genoomi järjestuse lõpetamiseks, sest universaalset, kõikidele
genoomidele sobivat lähenemisviisi, ei ole veel leitud. Sellise
programmi rakendamisel on kasutajatel vähe kontrolli
skäffoldimisprotsessi üle ja informatsiooni tootmise suunamist ei
saa kontrollida. Eraldiseisvad skäffolderid on paindlikumad ja
võimaldavad kontrollida skäffoldimisparameetrite rakendamist.
Esimene
täielikult lõpetatud elusorganismi genoom kuulub
Haemophilus
influenzaele, kelle
genoomi suuruseks on 1,89 Mb.
Shotgun
genoomi projekt hõlmab
endas mitut järku: DNA murdmine juhuslikest punktidest, saadud
järjestuste sekveneerimine ja assambleerimisprogrammidega
esialgse molekuli taastamine kasutades selleks eelnevalt sekveneeritud
fragmentide liitmist. Skäffoldimist peetakse vajalikuks etapiks kogu
genoomi
shotgun
projekti jaoks, kuna võimaldab kõrgkvaliteedilise mustandgenoomi
järjestuse loomist, millelelt edasiselt on võimalik andmete edasine
analüüs ja tulemuste tõlgendamine.
Joonis
1. Genoomi koostu paneb genoomi kokku lühikestest DNA
sekventsijuppidest.
Koostu
kvaliteetKuigi
esineb teravaid probleeme koostute tegemisel, ei takista see asjaolu
uute genoomiprojektide alustamist. Projekt i5k alustati aastal 2009
ja selle eesmärgiks on 5000 putukagenoomi sekveneerimine. Sellele
järgneb Genome 10K projekt, mille eesmärk on sekveneerida 10 000
selgroogse genoomi. Ulatuslikuma eesmärgi on seadnud Hiinas asuv
globaalne sekveneerimiskeskus BGI
Shenzhen,
mille sooviks on sekveneerida nii miljoni inimese, taime, mikroobi
kui ka looma genoomi.
Põhjused, miks koostus vead esinevad, on
mitmesugused. Järjestusetükid on koostu ülesehitamisel kõrvale
heidetud (märgistatud kui viga või kordus), tükkide ühinemine on
toimunud vales
asukohas või tükkide suund on vääralt määratud.
Lugemite ühendamisel saadud kontiigid on tihtipeale lühemad, kui
nad võiksid olla. Seda põhjustavad koostute jaoks
raskemad piirkonnad
genoomis : kordusjärjestused, polümorfismid. Samuti
teatud piirkonna andmete puudumine ja vead lugemites mõjutavad
kontiigide moodustamist.
Genoomide
hindamine toimub enamasti skäffoldide ja kontiigide arvu alusel.
Teatud piisav arv pikemaid järjestusi on vajalik genoomi
esindamiseks. Samuti kasutatakse nende järjestuste absoluutset ja
pikkust kogu genoomi suhtes.
Koostu
kvaliteeti hindamiseks on enimlevinud N50 suurus, mida määratletakse
kui kontiigi vähimat pikkust, millest võrdsed või pikemad
kontiigid katavad 50% kogu genoomsest järjestusest. Seega
laialdasemalt kasutusel olevad meetrikud
hindavad koostuid kontiigide
suuruse, mitte kontiigide kvaliteedi ja täpsuse alusel, mis
kajastaksid paremini koostu väärtust. Ebaõnnestunud koostute hulka
kuulub esialgne mantellooma genoom. Hilisem koostu laiendas N50
väärtust kümnekordselt ja paljastas konserveerunud geene, mida
varasemas koostus
esindatud polnud. Sama selgus
kana genoomse koostu
uurimisel.
Vajalik
on võrrelda erinevaid programme
samade andmete alusel. Iga uue
programmi kirjeldamisel, jooksutatakse seda kasutades konkreetseid
andmeid ehk omavahel on keeruline programme võrrelda, kui igaüks
jooksutab erineva andmete alusel. Assemblathon 1 ja 2,
GAGE (Genome
Assembly
Gold -standard Evaluations) ja dnGASP (de novo Genome
Assembly Assesment Project) on projektid, mille raames erinevad
tarkvara
arendajad moodustasid võistkondi ja proovisid enda
programmiga saavutada häid tulemusi. Töötlemiseks kasutatud andmed
olid selleks spetsiaalselt
stimuleeritud andmestik mitte päris
genoomid . Andmete valikut on kritiseeritud Assemblathon 1 puhul, kuna
tehislikud andmed ei suuda kunagi peegeldada
tegelikku olukorda
piisavalt rahuldaval tasemel. Assemblathon 2
projektis pidi töötlema
ka tõelisi sekveneerimisandmeid, mis pärinesid näiteks papagoilt
ja boamaolt.
JäreldusedAssemblathon,
GAGE ja dnGASP jõudsid kõik ühele järeldusele – ka kõige
paremad koostud sisaldavad olulisi ja rohkelt vigu. Iga bioloog peaks
silmas
pidama , et täiuslikku genoomi ei saa nendega koostada. Samuti
järeldus, et ideaalset ainuõiget meetrikut ei eksisteeri.
Stateegiad, mis teevad vähem vigu, teevad seda pikkuse arvelt. See
halvendab edasise analüüsi kvaliteeti, sest informatsiooni on vähe.
Võimalik oleks tulevikus jooksutada programme varasemalt
hästikirjeldatud genoomide (
hiir ja inimene) põhjal. Samuti peab
silmas pidama, et programm tuleb valida vastavalt andmete
spetsiifikale: see, mis sobib hästi prokarüootsete genoomide puhul,
ei pruugi sarnaseid tulemusi eukarüoodsete organismide korral
saavutada.
Selleks,
et paraneks koostute kvaliteet, tuleb tõsta sekveneerimisprotsessi
kvaliteeti. Samuti annab paremaid tulemusi, kui kasutada
lisainformatsiooni näiteks transkribeeritud RNA-d koostute
tegemisel. Kui tundub, et mingi piirkond genoomis on kehvasti
taaskonstrueeritud, võib seda parandada kasutades suunatud
sekveneerimist. Igatahes on vajalik edasine meetrikute analüüs,
töötada uusi välja ja leida spetsiaalseid meetrikuid ka raskemate
genoomsete piirkondade jaoks.
Kasutatud
kirjandusFleischmann,
R.D.,
Adams ,M.D., White, O.,
Clayton , R.A., Kirkness, E.F.,
Kerlavage, A.R., Bult, C.J.,
Tomb , J.F., Dougherty, B.A., Merrick,
J.M., et al. (1995).
Whole -genome
random sequencing and assembly of
Haemophilus influenzae Rd. Science 269 (5223): 496-512.
International
Human Genome Sequencing Consortium. (2004).
Finishing the euchromatic
sequence of the human genome.
Nature 431: 931-945.
Godson,
G.N, Barrell, B.G., Staden, R., Fiddes, J.C. (1978). Nucleotide
sequence of bacteriophage G4 DNA. Nature 276: 236-247.
Gritsenko,
A.A, Nijkamp, J.F., Reinders, M.J.T, de Ridder, D. (2012).
Bioinformatics 28(11): 1429-37.
Narzisi
G., Mishra, B. (2011). Comparing de novo genome assembly: the long
and short of it. PLoS One. 6 (4).
Wu,
W., Stupi, B.P., Litosh, V.A., Mansouri, D., Farley, D.,
Morris , S.,
Metzker, S., Metzker, M.L. (2007). Termination
of DNA
synthesis by N6-alkylated,
not 3′-O-alkylated,
photocleavable 2′-deoxyadenosine triphosphates. Nucleic Acids
Research, 35 (19): 6339-6349.
Xue,
W., Li, J.-T., Zhu, Y.-P., Hou, G.-Y.,
Kong , X.-F., Kuang, Y.-Y.,
Sun, X.-W. (2013). L_RNA_scaffolder:
scaffolding genomes with transcripts
L_RNA_scaffolder:
scaffolding genomes with transcripts
L_RNA_scaffolder:
scaffolding genomes with transcripts
L_RNA_Scaffolder:
scaffolding genomes with transcripts. BMC Genomics 14: 604.
Zhang ,
J., Chiodini, R., Badr, A., Zhang, G. (2011). The impact of
next-generation sequencing on genomics. J Genet Genomics 38 (3):
95-109.
























Kõik kommentaarid